论文通讯作者、Genetech首席科学家—— Shiva Malek
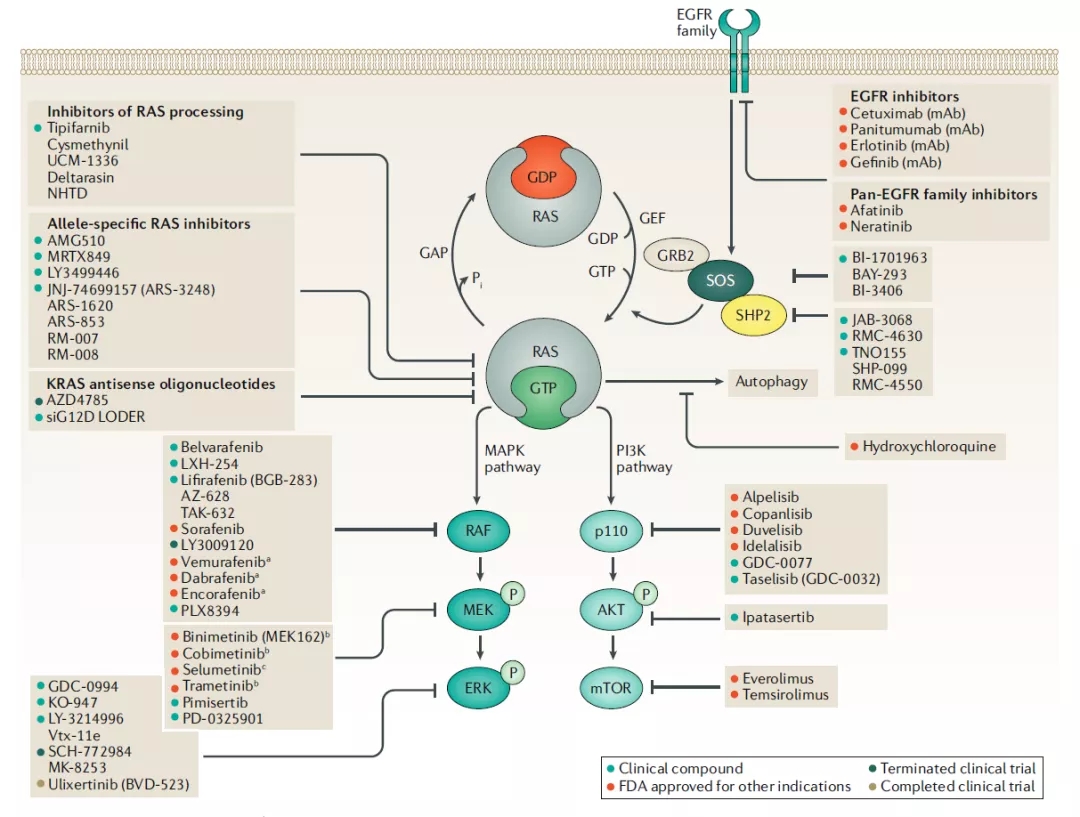
RAS突变肿瘤抑制剂的临床开发
RAS突变和剪接变体
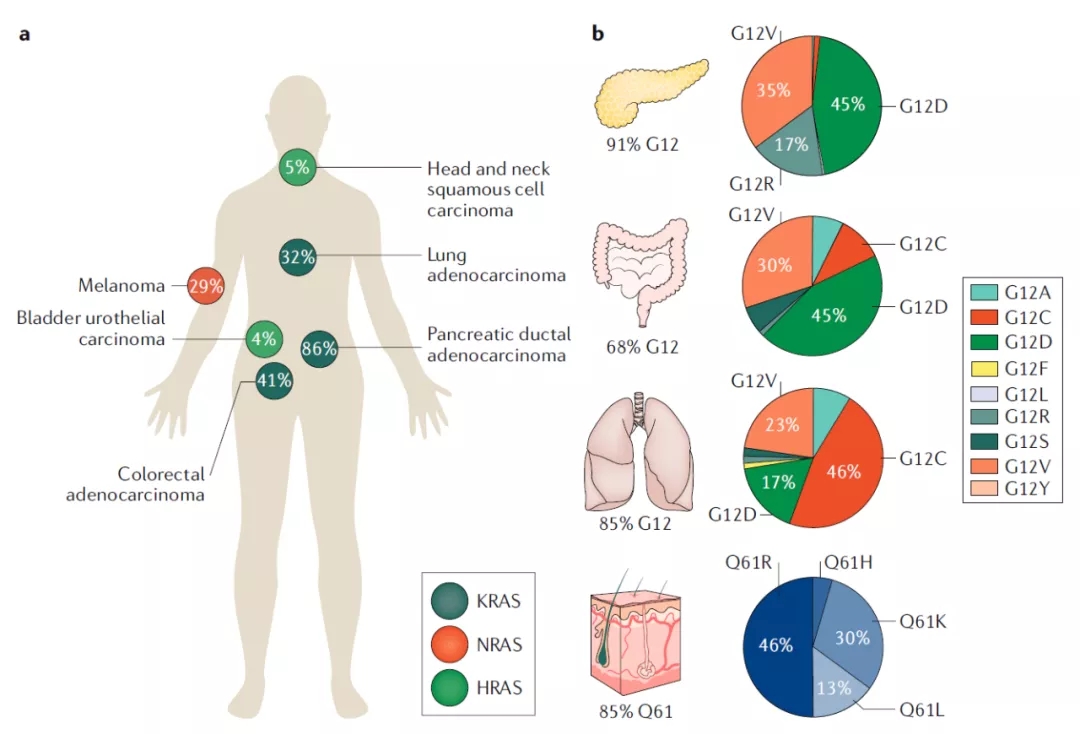
KRAS基因编码时使用不同4号外显子,从而产生两种剪接变体—— KRAS4A和KRAS4B。长期以来,KRAS4B被认为是主要的剪接变体,因为它在人类癌症中普遍存在并且高度表达。
直接靶向RAS
直接靶向抑制RAS是治疗RAS突变肿瘤的理想方法,直接靶向RAS包括switch-II突变体选择性共价抑制剂和pan-RAS抑制剂。
靶向KRAS-G12C的共价抑制剂
KRAS中G12是突变热点,而G12C是该位置的第三大最常见突变,共价靶向活性位点半胱氨酸是药物发现中广泛使用的策略,KRAS-G12C的半胱氨酸可以被用来开发共价小分子抑制剂,重要的是,野生型KRAS在活性位点缺乏半胱氨酸,因此可以通过这种方法特异性抑制KRAS-G12C。
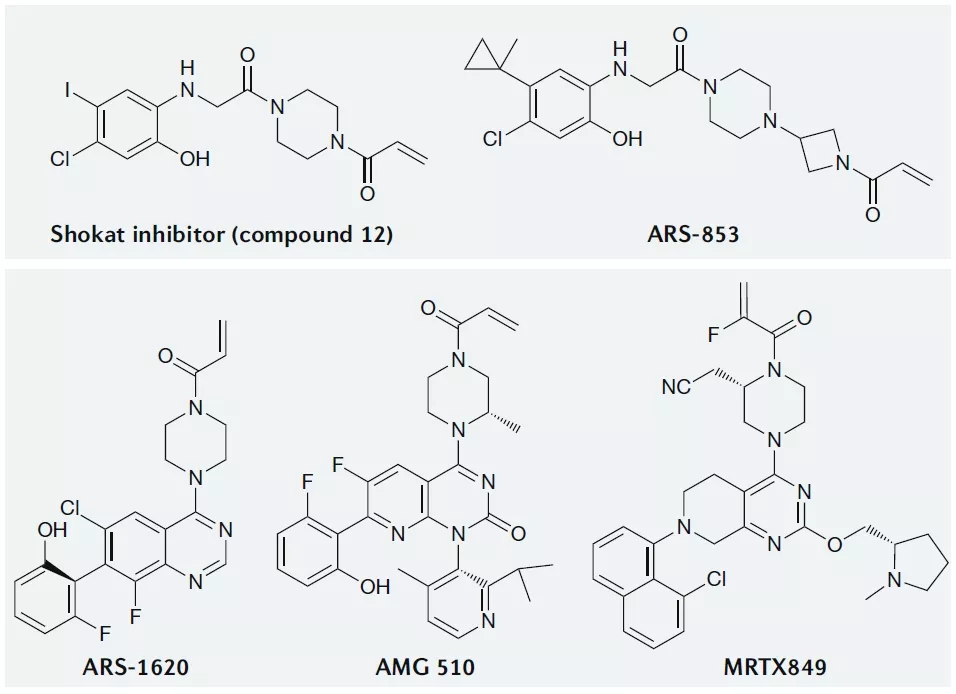
靶向KRAS-G12C的小分子抑制剂
间接靶向RAS的方法
正常的RAS激活需要核苷酸交换,加工,膜定位和效应子结合。改变这些基本步骤中的一个,可用于间接抑制RAS激活。
核苷酸交换循环抑制剂
1、SOS抑制剂
SOS1抑制剂可与结合GDP状态的KRAS-G12C抑制剂结合使用,因为SOS1抑制会增加GDP结合的KRAS-G12C的总量。目前,SOS1抑制剂BI-1701963作为一种单一药物,以及与MEK抑制剂曲美替尼的组合疗法正处于I期临床试验中。
2、SHP2抑制剂
源自临床候选药物的RMC-4550,RMC-4630正在进行单药I期临床试验,以及和与MEK抑制剂考比替尼组合的Ib/II期临床试验。
第二种SHP2变构抑制剂JAB-3068目前正在进行I/II期临床试验,第三种SHP2抑制剂TNO155正在进行I期单药临床试验,以及与MRTX849组合的I/II期与临床试验。目前这些临床试验结果都还未公布。
RAS加工的抑制剂
RAS蛋白仅在定位于细胞膜时才有活性。为了与细胞膜膜结合,RAS需要进行三个酶促反应,因此抑制RAS的加工可以防止其与细胞膜结合及下游RAS信号传转导。
但值得注意的是,抑制这些与RAS加工有关的酶,可能导致脱靶效应。因此,直接抑制RAS最有可能成为主要的治疗选择。
RAS寡聚和效应子结合
最近发现的RAS自动抑制新机制——膜闭塞,为靶向RAS蛋白-蛋白相互作用提供了另一种潜在途径。在膜闭塞中,RAS与脂质膜之间的直接相互作用将RAS的效应子结合界面从细胞质中分离出来。
一个小分子Cmpd2可以结合到RAS和脂质膜之间的界面(Kd=1μM),促进膜阻塞并减少与RAF的RBD结构域的结合。
由于RAS-效应子接口的高度保守性,这种类型的策略为有效抑制RAS驱动的所有下游信号通路提供了机会。
靶向RAS通路
有两种靶向RAS通路的不同方法:鉴定具有RAS突变致死性的基因,靶向酪氨酸激酶受体(EGFR家族)或RAS效应子通路,即MAPK和PI3K。
1、合成致死筛选
在KRAS突变和NRAS突变细胞系中,除了RAS本身外,最重要的必需基因是RAF1(编码CRAF)和SHOC2。RAS突变的人类急性髓细胞白血病细胞株需要RAF1和SHOC2。
2、EGFR家族疗法
有证据表明RAS与受体酪氨酸激酶的EGFR家族之间存在相互作用。由于EGFR家族位于RAS的上游,因此这些受体酪氨酸激酶的失活可以减少RAS的激活。
3、MAPK途径:RAF抑制剂
活性GTP结合的RAS促进RAF二聚化和磷酸化,从而诱导RAF激酶活性并导致RAF底物MEK1和MEK2磷酸化。磷酸化级联继续与末端激酶ERK1和ERK2的MEK磷酸化。
ERK激酶活性既激活包括ETS家族成员在内的促进生长的转录因子,又参与负反馈回路,此动态过程控制MAPK信号级联的持续时间和幅度。
ERK可以通过磷酸化上游成分SOS或CRAF或改变双特异性磷酸酶(DUSP)家族和Sprouty(SPRY)家族的转录来负面抑制MAPK信号传导。DUSPs通过隔离SOS-GRB2来使ERK和SPRYs磷酸化,从而抑制受体酪氨酸激酶信号传导。
为了有效治疗RAS突变肿瘤,必须几乎完全抑制MAPK途径。实现这一目标的一种策略是使用针对MAPK途径成分的激酶抑制剂,结合针对本综述中讨论的其他机制的疗法。
4、MAPK通路:MEK抑制剂
目前,有三种靶向MEK的变构激酶抑制剂——Cobimetinib、Trametinib和Binimetinib,临床批准用于治疗 BRAF V600 突变型黑色素瘤。但MEK抑制剂作为RAS突变肿瘤的单一疗法的临床试验没有发现任何改善,目前没有一种MEK抑制剂被临床批准用于治疗RAS突变肿瘤。
但是,在具有高水平内在核苷酸交换水平的突变KRAS细胞(例如KRAS-G13D)中观察到了MEK和RAF的最强协同作用,该突变KRAS增强了GTP结合的RAS水平:该观察结果表明, MEK抑制可导致RAF二聚化,可能有助于这种协同作用。
目前,有两项RAF抑制剂和MEK抑制剂联用的I期临床试验正在进行。一个Belvarafenib和Cobimetinib的联用,另一个是曲美替尼和LXH-254的联用。
5、MAPK通路:ERK抑制剂
抑制级联反应中的最终激酶ERK可以直接降低致癌转录产物,并为抗MEK或RAF抑制作用的肿瘤提供有价值的治疗选择。ERK抑制剂的临床开发落后于MEK,BRAF单体和pan-RAF抑制剂的开发。
此外,ERK抑制剂用于治疗RAS突变肿瘤的早期临床试验在很大程度上还没有成功。
总体而言,单药MEK、RAF或ERK抑制剂在治疗RAS突变型肿瘤方面几乎没有疗效。因此,这些抑制剂将必须与MAPK通路的其他抑制剂或本综述中讨论的其他方法结合使用。寻找每种抑制剂的最佳组合将是一个挑战,但是,等位基因特异性RAS抑制剂的出现增加了可用于实现最大途径抑制的潜在组合的数量。
6、PI3K通路抑制剂
在所有RAS效应子中,MAPK通路一直是抑制RAS突变型肿瘤的主要焦点。但是,RAS也激活了第二个效应子通路PI3K。
目前有四种I类PI3K抑制剂获得FDA批准,但这些抑制剂都未被批准用于治疗RAS突变肿瘤。RAS激活PI3K和MAPK通路,并且存在重叠的反馈机制。抑制一个通路可导致另一种通路的代偿性激活。因此,同时抑制MAPK和PI3K是一种值得注意的策略。
临床前研究表明,PI3K和MEK的联合抑制对于RAS突变肿瘤的治疗是有效的,在临床上是可以实现的。
然而,在临床试验中,这些抑制剂的组合是不耐受的,因此几乎没有实际疗效,大概是由于毒性导致剂量减少的结果。
临床中的单药治疗

临床中的组合疗法

新兴疗法
小干扰RNA(siRNA)疗法
阿斯利康开发的一款名为AZD4785的siRNA疗法,在小鼠模型中能够有效特异性靶向KRAS突变,但临床试验结果并不显着。
而Silenseed公司进行的名为siG12D- LODER的siRNA疗法,在1期临床试验中显示出对胰腺导管腺癌(PDAC)的希望,而与标准化学疗法联用的2期临床试验正在进行中。
自噬
胰腺导管腺癌(PDAC)癌细胞的细胞自噬水平升高,这些细胞需要自噬才能生长,羟氯喹能够有效抑制自噬水平,尽管用羟氯喹治疗PDAC的疗效有限,但是在NRAS突变型黑色素瘤和PDAC的临床前模型中,羟氯喹和MAPK抑制剂的组合显示出令人鼓舞的结果。
有了这个令人鼓舞的结果,目前正在评估一项1期临床试验,以研究曲美替尼联合羟氯喹治疗胰腺导管腺癌(PDAC)患者的疗效。
免疫疗法
1、免疫检查点抑制剂
有研究表明,KRAS突变的肿瘤中PD-L1水平升高,以提高免疫逃逸。目前已有多项RAS抑制剂、MEK抑制剂、SHP2抑制剂与PD-1/PD-L1抑制剂联用治疗非小细胞肺癌的临床试验。
2、过继细胞疗法
治疗RAS驱动癌症的第二种免疫治疗方法涉及改造免疫系统,以特异性识别突变RAS蛋白。这种方法通过改造T细胞,离体扩增后再回输到患者体内。目前有两项分别靶向RAS-G12D和RAS-G12V的过继细胞疗法临床试验正在进行中。
3、癌症疫苗
治疗RAS突变肿瘤的第三种免疫疗法方法是使用已知的RAS突变肿瘤抗原通过疫苗接种引起T细胞应答。
未来发展方向
KRAS-G12C等位基因特异性抑制剂将改变RAS驱动肿瘤的治疗前景。这些抑制剂有望成为FDA批准的RAS突变型肿瘤的首批治疗剂,并将用于治疗由RAS突变驱动的难治性癌症,例如PDAC,CRC和LUAD。尽管这些抑制剂的开发振奋人心,但新的挑战和问题也将出现。
首先,RAS中的每个变异都有独特的生化特性,这些特性将决定对治疗方法的不同反应。例如,使用RMC-4550抑制SHP2可以有效治疗具有KRAS G12突变的细胞,并且对KRAS G12C比对KRAS G12D或KRAS G12V更有效。该观察结果表明,SHP2和KRAS-G12C抑制剂的组合将是一种有效的治疗策略。在开发联合疗法时,有必要了解特定突变体RAS密码子的要求和抑制剂对这些等位基因的反应。
其次,肿瘤类型会极大地影响缓解率,RAS突变的CRC和PDAC对MAPK抑制剂或免疫检查点封锁的反应很小。AMG 510试验的早期数据表明,CRC比LUAD更难治疗,表明CRC需要使用联合疗法。
第三,第三,从经验来看,联合疗法容易有毒副作用,且安全性较差。然而,AMG 510并未观察到剂量限制性毒性,并且突变体特异性疗法应具有有限的脱靶作用。等位基因特异性抑制剂(毒性低)可以与毒性更大的其他抑制剂组合使用,而不会遇到像PI3K和MEK抑制剂组合那样将两种有毒化合物组合在一起所观察到的问题。
总的来说,FDA对RAS靶向疗法AMG510开放快速通道,这是一个激动人心的时刻,让我们对成功治疗RAS突变癌症重新燃起了希望。
参考链接:
https://www.nature.com/articles/s41573-020-0068-6
扫描上面二维码在移动端打开阅读