癌症免疫监测(Cancer immunosurveillance)主要依赖传统 CD8 T 细胞识别肿瘤新抗原(Neoantigen)进而杀伤癌细胞。然而癌细胞能通过上调具有免疫抑制功能的 PD-L1 与传统 CD8 T 细胞中的 PD-1 受体相互作用,使 T 细胞的效用功能减少,甚至丧失,从而诱导 T 细胞耗竭(T cell exhaustion)。
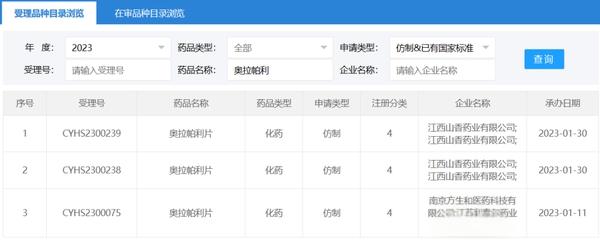
近十年,利用靶向抗体抑制 PD-1 或 PD-L1 来恢复传统 T 细胞效用功能的免疫疗法已经在临床上取得一定的成效,并因此荣获2018诺贝尔生理医学奖。至此,研发更多针对传统 T 细胞的检查点抑制剂成为肿瘤免疫疗法的主导思路。
然而,尽管研究人员和临床肿瘤专家对免疫检查点抑制疗法抱有很大的希望,其疗效却往往受制于肿瘤新抗原量。因此不是所有类型的癌症都适用,并且只有少部分患者能受惠于该疗法。利用单细胞转录组测序技术,许多团队发现在小鼠模型与癌症患者的肿瘤组织中其实充斥着种类繁多的肿瘤浸润 T 细胞。然而,除了高表达 PD-1 的 CD8 T 细胞外,我们对于其他 T 细胞在癌症免疫监测中的作用却知之甚少。
2022年4月20日晚23时,美国纪念斯隆凯特琳癌症中心李明教授团队(周骏博士、张晛博士为共同第一作者)在 Nature 期刊发表了题为:Programme of self-reactive innate-like T cell-mediated cancer immunity 的研究论文,该研究揭示了一种免疫抗癌新机制。
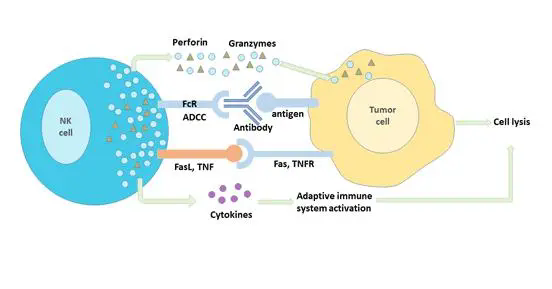
在最新这项研究中,研究团队首先在小鼠乳癌模型(PyMT)发现一种缺乏 PD-1 却高表达自然杀伤细胞(Natural Killer Cell)受体的类先天性杀伤型 T 细胞(Killer Innate-like T cell, ILTCK)。虽然 ILTCK 和传统 CD8 T 细胞都表达 T 细胞受体(T cell receptor,TCR),前者的激活却不依赖树突细胞(Dendritic Cell,DC),此特性使其更接近先天性淋巴细胞(Innate lymphoid cell)。不同于传统 CD8 T 细胞,ILTCK 不表达 PD-1 和其他免疫抑制受体,因此不会进入细胞耗竭状态,反而对肿瘤细胞有更强大的细胞毒性(cytotoxicity)。其次,大部分的传统 T 细胞识别肿瘤新抗原,而 ILTCK 识别肿瘤原生抗原,并且具有显著的组织驻留性(tissue residency)。
研究团队发现 ILTCK 并非分化于初始(naïve)CD8 T 细胞,而是独立于传统 T 细胞的另一细胞谱系。在 T 细胞分化过程中,绝大部分能够识别宿主原生抗原的T细胞通常能被有效地移除进而避免自体免疫疾病。然而 ILTCK 识别自体原生抗原的特性却是该细胞谱系分化的关键信号,但此分化机制能够永久抑制 ILTCK 中 T 细胞受体的下游信号传递。因此,广泛存在的 ILTCK 并无法引发自体免疫疾病,反而能够在原生抗原遍布的肿瘤内免于细胞耗竭。
李明团队进一步揭示白细胞介素15(IL-15)为癌细胞激活 ILTCK 的关键他们发现正常组织几乎不表达 IL-15,然而癌症细胞上调该信号。在癌症细胞内敲除 IL-15 大幅降低肿瘤浸润 ILTCK 数量及其效用功能,导致肿瘤快速生长。反之,在 ILTCK 内增强 IL-15 信号传递能够有效地抑制肿瘤发生。因此,IL-15 是一种新型的危险素(alarmin):ILTCK 透过监测该信号的上调进而识别并歼灭机体中潜在的癌症细胞。
ILTCK 的发现为肿瘤免疫疗法提供了一个新的选择。此类细胞在组织驻留性和抗细胞耗竭的能力上都优于传统 CD8 T 细胞(图1)。现有的T细胞输入疗法,如 CAR-T 等,大部分选择传统 T 细胞作为基底。虽然在治疗血癌上颇有成效,但传统 T 细胞的弱组织驻留性和易于耗竭的弱点使该疗法对实体肿瘤束手无策。因此,ILTCK 作为基底的 T 细胞输入疗法或许能更有效的抑制实体肿瘤。
最后,虽然此研究大部分着重于小鼠模型,但研究团队在人类大肠癌患者的组织中也发现类似于 ILTCK 的 T 细胞 (图2)。ILTCK 的临床转化将是研究团队的下一个研究重点。
论文链接:
https://www.nature.com/articles/s41586-022-04632-1
近十年,利用靶向抗体抑制 PD-1 或 PD-L1 来恢复传统 T 细胞效用功能的免疫疗法已经在临床上取得一定的成效,并因此荣获2018诺贝尔生理医学奖。至此,研发更多针对传统 T 细胞的检查点抑制剂成为肿瘤免疫疗法的主导思路。
然而,尽管研究人员和临床肿瘤专家对免疫检查点抑制疗法抱有很大的希望,其疗效却往往受制于肿瘤新抗原量。因此不是所有类型的癌症都适用,并且只有少部分患者能受惠于该疗法。利用单细胞转录组测序技术,许多团队发现在小鼠模型与癌症患者的肿瘤组织中其实充斥着种类繁多的肿瘤浸润 T 细胞。然而,除了高表达 PD-1 的 CD8 T 细胞外,我们对于其他 T 细胞在癌症免疫监测中的作用却知之甚少。
2022年4月20日晚23时,美国纪念斯隆凯特琳癌症中心李明教授团队(周骏博士、张晛博士为共同第一作者)在 Nature 期刊发表了题为:Programme of self-reactive innate-like T cell-mediated cancer immunity 的研究论文,该研究揭示了一种免疫抗癌新机制。

在最新这项研究中,研究团队首先在小鼠乳癌模型(PyMT)发现一种缺乏 PD-1 却高表达自然杀伤细胞(Natural Killer Cell)受体的类先天性杀伤型 T 细胞(Killer Innate-like T cell, ILTCK)。虽然 ILTCK 和传统 CD8 T 细胞都表达 T 细胞受体(T cell receptor,TCR),前者的激活却不依赖树突细胞(Dendritic Cell,DC),此特性使其更接近先天性淋巴细胞(Innate lymphoid cell)。不同于传统 CD8 T 细胞,ILTCK 不表达 PD-1 和其他免疫抑制受体,因此不会进入细胞耗竭状态,反而对肿瘤细胞有更强大的细胞毒性(cytotoxicity)。其次,大部分的传统 T 细胞识别肿瘤新抗原,而 ILTCK 识别肿瘤原生抗原,并且具有显著的组织驻留性(tissue residency)。
研究团队发现 ILTCK 并非分化于初始(naïve)CD8 T 细胞,而是独立于传统 T 细胞的另一细胞谱系。在 T 细胞分化过程中,绝大部分能够识别宿主原生抗原的T细胞通常能被有效地移除进而避免自体免疫疾病。然而 ILTCK 识别自体原生抗原的特性却是该细胞谱系分化的关键信号,但此分化机制能够永久抑制 ILTCK 中 T 细胞受体的下游信号传递。因此,广泛存在的 ILTCK 并无法引发自体免疫疾病,反而能够在原生抗原遍布的肿瘤内免于细胞耗竭。
李明团队进一步揭示白细胞介素15(IL-15)为癌细胞激活 ILTCK 的关键他们发现正常组织几乎不表达 IL-15,然而癌症细胞上调该信号。在癌症细胞内敲除 IL-15 大幅降低肿瘤浸润 ILTCK 数量及其效用功能,导致肿瘤快速生长。反之,在 ILTCK 内增强 IL-15 信号传递能够有效地抑制肿瘤发生。因此,IL-15 是一种新型的危险素(alarmin):ILTCK 透过监测该信号的上调进而识别并歼灭机体中潜在的癌症细胞。
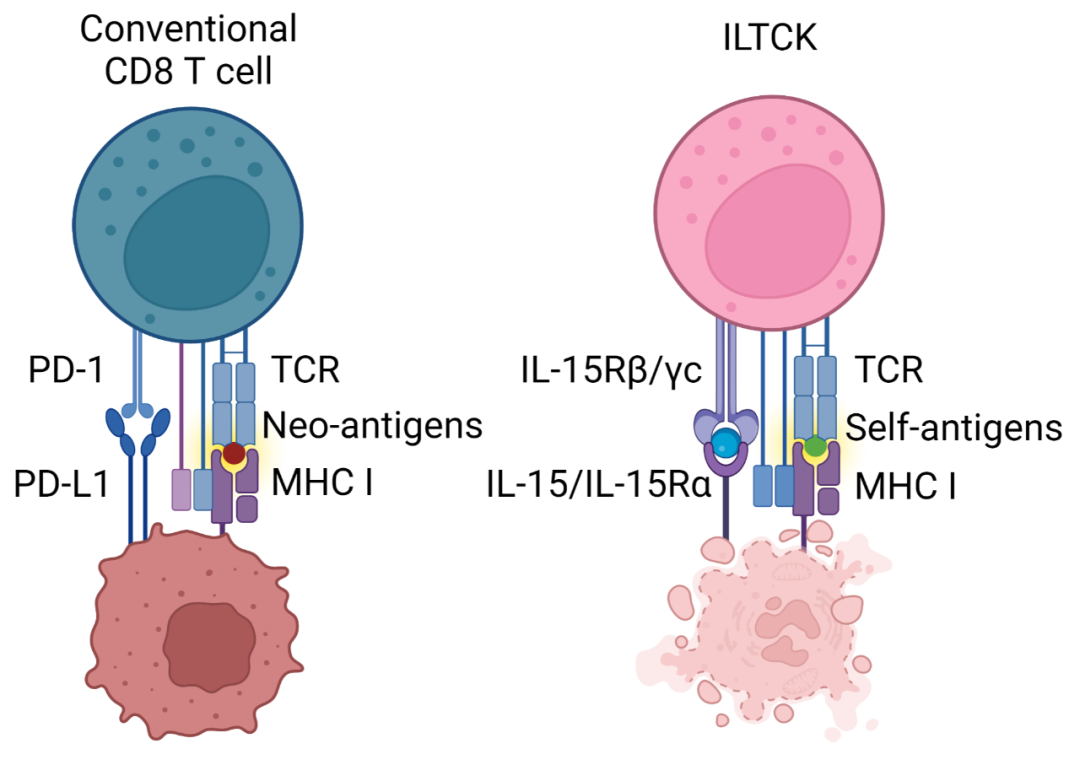
ILTCK 的发现为肿瘤免疫疗法提供了一个新的选择。此类细胞在组织驻留性和抗细胞耗竭的能力上都优于传统 CD8 T 细胞(图1)。现有的T细胞输入疗法,如 CAR-T 等,大部分选择传统 T 细胞作为基底。虽然在治疗血癌上颇有成效,但传统 T 细胞的弱组织驻留性和易于耗竭的弱点使该疗法对实体肿瘤束手无策。因此,ILTCK 作为基底的 T 细胞输入疗法或许能更有效的抑制实体肿瘤。
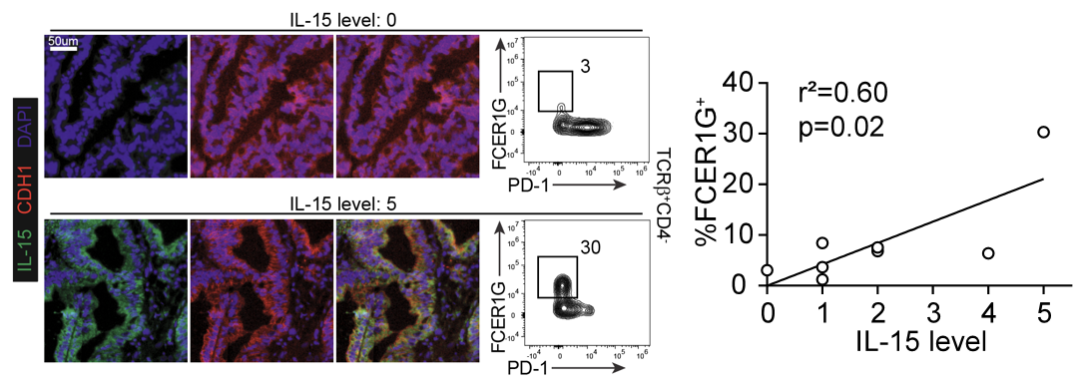
最后,虽然此研究大部分着重于小鼠模型,但研究团队在人类大肠癌患者的组织中也发现类似于 ILTCK 的 T 细胞 (图2)。ILTCK 的临床转化将是研究团队的下一个研究重点。
论文链接:
https://www.nature.com/articles/s41586-022-04632-1
扫描上面二维码在移动端打开阅读