
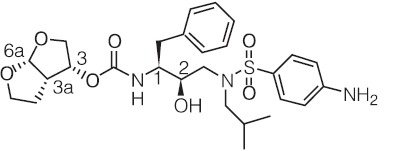
药品概述
darunavir是杨森开发的一种蛋白酶抑制剂,在欧盟以商品名Prezista上市,该药常与一种增效剂ritonavir及其他HIV药物联合用药。
主要成份:达芦那韦
本品为白色薄膜衣异形片,除去包衣后显白色或类白色。
达芦那韦联合100mg利托那韦合用(达芦那韦/利托那韦),和其他抗逆转录病毒药物合并使用,适用于已使用过抗逆转录病毒药物的HIV(人类免疫缺陷病毒)感染的成人患者的治疗,例如对一种以上蛋白酶抑制剂耐药的HIV-1感染者。
这一适应症基于两项达芦那韦/利托那韦与其他抗病毒药物联合的对照试验中血浆HIV RNA水平与CD4+细胞计数24周分析结果。两项试验的受试者均为临床晚期、经历抗病毒治疗(NRTIs、NNRTIs和Pis)的成人患者,这些患者尽管在进行抗病毒治疗但未能抑制HIV-1复制。
对于初治患者或儿童患者,达芦那韦/利托那韦的风险和收益缺乏相关资料。
300mg。
使用方法:口服。
使用达芦那韦时,必须以100mg利托那韦作为药代动力学增效剂,并联合其它抗逆转录病毒药物。因此在开始达芦那韦/利托那韦治疗前,必须参考利托那韦的处方资料。
成人:达芦那韦的推荐剂量是每次60mg,每天2次(b.i.d.),与利托那韦(每次100mg,每天2次)及食物同服。食物的类型不影响达芦那韦的吸收。利托那韦(每次100mg,每天2次)用来作为达芦那韦的药代动力学增效剂(见药物相互作用和药代动力学部分)。进一步增加达芦那韦或利托那韦的剂量不太可能引起临床抗病毒活性的相应增加。
儿童患者:达芦那韦在儿童患者中的安全性和有效性尚未通过验证。
肝脏损害:对于肝功能损害的患者,目前尚无使用达芦那韦/利托那韦的相关资料,因此没有具体的推荐剂量。肝脏损害的患者应慎用达芦那韦/利托那韦(见注意事项及药代动力学部分)。
肾脏损害:对于中度肾功能障碍的患者,无需调整剂量,对于重度或终末期肾病的HIV-1感染患者,尚缺乏药代动力学数据(见注意事项及药代动力学部分)。
1.1在汇总的POWER 1、2和3中发现的本品/利托那韦的药物不良反应
在汇总的POWER试验中,立即开始使用本品/利托那韦600/100mg、b.i.d.治疗的患者的总暴露为812.4患者年。
在本品/利托那韦治疗期间所报告的大部分不良反应(ADR)均为轻度。
最常见的(≥5%)中重度(2-4级)ADR包括腹泻、头痛、腹痛、恶心和呕吐。
最常见的3级或4级ADR包括肝酶及胰酶升高、高甘油三酯血征、腹泻、高胆固醇血征、头痛、腹痛和呕吐。所有其他3级或4级ADR的发生率均低于1%。
2.1%的患者因ADR而终止了治疗。
下表总结了在汇总的POWER1、2和3试验中本品/利托那韦600/100mg b.i.d.用于未接受过抗逆转录病毒治疗的成年HIV-1患者时发生的至少为中度(2-4级)的药物不良反应*:
http://x1.webres.medlive.cn/drugref/ChemPreparationDetail/201511231118170424.jpg
下表总结了在汇总的POWER1、2和3试验中本品/利托那韦用于接受过抗逆转录病毒治疗的成年HIV-1患者中发生的被认为是ADR的实验检查结果异常:
http://x1.webres.medlive.cn/drugref/ChemPreparationDetail/201511231119590886.jpg
1.2在其他临床试验中发现的本品/利托那韦的其他药品不良反应:Artemis[TMC114-C211],Titan[TMC114-C214],TMC114-C208,TMC114-C209,DUET_1[TMC125-C206],DUET_2[TMC125-C216]或其他正在进行中的试验。
http://x1.webres.medlive.cn/drugref/ChemPreparationDetail/201511231120500069.jpg
在上述试验或其他正在进行的试验中,未发现其他ADR术语。
2 联合抗逆转录病毒治疗的效果
HIV患者中联合抗病毒治疗与身体脂肪重新分布(脂肪代谢障碍)相关,这些异常包括外周和面部皮下脂肪丢失,腹腔和内脏脂肪增多,乳房肥大,以及颈背部脂肪堆积(水牛背)。
联合抗病毒治疗与代谢异常相关,如高甘油三酯血征,高胆固醇血征,胰岛素抵抗,高血糖征及高乳糖血征。
在开始联合抗逆转录病毒治疗时存在严重免疫缺陷的HIV感染患者,可能会出现对无症状或残留的机会性感染的炎症反应。
接受蛋白酶抑制剂(PIs)治疗血友病患者中已有自发性出血增多的报告。
使用蛋白酶抑制剂,特别是与NRTIs联合使用时,已有CPK升高,肌痛,肌炎的报道,极少数患者可出现横纹肌溶解。
3特殊人群
乙型肝炎和/或丙型肝炎共感染的患者
在乙型肝炎或丙型肝炎共感染并接受本品/利托那韦的患者中,除肝酶升高外,不良反应和临床生化检查异常的发生率不高于无共同感染并接受本品/利托那韦治疗的患者。共感染患者与无共感染患者的药代动力学暴露量相当。对肝炎共感染患者给予标准的临床监测是可行的。
对达芦那韦或达芦那韦所含任何一种成份过敏者。
达芦那韦和利托那韦都是细胞色素P450 3A(CYP3A4)异构体的抑制剂。达芦那韦/利托那韦不应与高度依赖CYP3A4清除的药物同时服用,因为这些药物血浆浓度增高与严重和/或危及生命的事件相关(治疗指数狭窄)。这些药物包括阿司咪唑(息斯敏)、特非那丁、咪达唑仑、三唑仑、西沙比利、哌迷清和麦角生物碱(如麦角胺、双氢麦角胺、麦角新碱和甲基麦角新碱)(见药物相互作用部分)。
1.应告知患者目前的抗逆转录病毒治疗还不能治愈HIV,而且还没有证实能够预防HIV的传播。应继续使用适当的预防措施。
2.在考虑到对幼鼠给药(20mg/kg到1,000mg/kg)23到26天观察到毒性,本品/利托那韦不能用于3岁以下儿童,(见药理毒理部分)
本品/利托那韦的安全性和有效性尚未在对已接受过抗逆转录病毒治疗的3-6岁儿童和未接受过抗逆转录病毒治疗的儿童患者中确定。
3.老年人:因在65岁及以上的患者中使用达芦那韦的资料有限,因此老年患者应慎用达芦那韦,有肝功能降低及伴随疾病或其它治疗增加的可能性(见药代动力学部分)。
4.单次剂量600mg本品的绝对口服生物利用度大约是37%,与100mg利托那韦b.i.d.合用时,增加到约82%,利托那韦的药代动力学增强效果大约可使达芦那韦的全身暴露量增加14倍。因此,本品应与低剂量的利托那韦联合使用(见药代动力学部分)。
增加利托那韦的剂量对达芦那韦的浓度无明显影响,因此不推荐增加剂量。
5.在临床试验中(n=3063),皮疹(所有级别,无论原因为何)在接受PREZISTA治疗的患者中的发生率为10.3%(见不良反应部分)。大部分皮疹均为轻至中度,常常发生在治疗的前四周内,在不停药的情况下都可以治愈。在接受本品/利托那韦治疗的患者中,因皮疹而 停药的患者占0.5%。
严重的皮疹(可能伴有发热和/或转氨酶升高)在患者中的发生率为0.4%。多形性渗出性红斑罕见(<0.1%)。如果出现了严重的皮疹,应停用本品。
6.达芦那韦含有磺胺。在已知对磺胺过敏的患者中,应慎用本品。在一项针对本品/利托那韦的临床研究中,有磺胺过敏史的患者中皮疹的发生率和严重程度与无磺胺过敏史的患者是相当的。
7.请置于儿童不易拿到处
伴随疾病的患者
肝脏损伤
目前尚无严重肝损伤患者使用本品/利托那韦的相关资料;因此,不推荐具体剂量。严重肝损伤患者应慎用本品/利托那韦。试验数据证实,轻度或中度肝损伤受试者使用达芦那韦时,其稳态药代动力学参数与健康受试者的参数接近,轻度或中度肝损伤患者无需进行剂量调整(见用法用量和药代动力学部分)。
曾患肝功能障碍的病人,包括慢性活动性乙型或丙型肝炎,在联合抗逆转录病毒治疗中肝脏功能异常的发生频率可能增加,应根据标准程序进行监测。在这些病人中,如果有肝脏疾病恶化的证据,需要考虑中断或中止治疗。
肾脏损伤
因为达芦那韦的肾脏清除有限,在肾脏损害的病人中,机体总的清除率预期不会降低。达芦那韦和利托那韦与血浆蛋白高度结合,因此它们不太可能通过血液透析或腹膜透析而大量清除(见用法用量和药代动力学部分)。
血友病患者
在使用PIs治疗的A型和B型血友病患者中,已有出血增加的报道,包括自发的皮肤血肿和关节血肿。在部分病人中给予了额外的VIII因子。半数以上的报告病例中,如果已经停止治疗,可继续或再引入PIs治疗。尽管这种导致出血的作用机制还没有被阐明,但已提示因果关系。因此,应使血友病患者意识到出血增加的可能性。
高血糖征
在接受抗逆转录病毒治疗(包括PIs治疗)的病人中,已经有新发糖尿病,高糖血征或糖尿病恶化的报道。部分病人中,高糖血征很严重,部分病例尚会出现酮症酸中毒。许多病人的病情较为复杂,其中一些情况需要药物治疗,但这些药物又可引起糖尿病或高血糖征进一步恶化。
脂肪重新分布&代谢紊乱
在HIV感染病人中,联合抗逆转录病毒治疗可引起机体脂肪的重新分布(脂肪代谢障碍)。这些副作用的长期后果目前尚不明确,对其机制的了解还不完善。目前认为内脏脂肪过多与PIs、皮下脂肪萎缩和NRTIs有一定关系,脂肪代谢障碍的高危因素包括个体因素如年龄较大,药物相关因素如抗病毒治疗持续时间较长,以及与治疗有关的代谢障碍。临床上应对脂肪重新分布的体征进行评估,检测空腹血脂和血糖,对脂质紊乱给予适当的处理(见不良反应)。
免疫重建炎性综合征
严重免疫缺陷的艾滋病患者在开始联合抗病毒治疗(CART)后,可能出现对无症状或残余的机会性病原菌的炎性反应,并引起严重的临床疾病,或症状好恶化。一般在开始CART的前几周或数月内可以观察到这种反应。有关的例子如巨细胞病毒性视网膜炎、播散性和/或局灶性分枝杆菌感染、及肺孢子虫性肺炎。对任何炎性症状应进行评估,必要时给予治疗。
药物相互作用
达芦那韦和利托那韦都是CYP3A4异构体的抑制剂。达芦那韦和利托那韦与主要由CYP3A4代谢的药物同时服用时,可导致这些药物血浆浓度增加,从而增加或延长它们的疗效和副作用(见禁忌和药物相互作用部分)。
达芦那韦经CYP3A4代谢。能够诱导CYP3A4活性的药品可能会增加达芦那韦的清除率,从而降低达芦那韦的血浆浓度(见药物相互作用部分)。
达芦那韦与其他能够抑制CYP3A4活性的药品合用时,可能会降低达芦那韦的清除率,从而增加达芦那韦的血浆浓度(见药物相互作用部分)。
妊娠
达芦那韦在妊娠妇女中的研究较少,也没有较好的对照研究。动物试验中没有发现发育毒性或影响生殖和生育功能的证据(见毒理研究部分)。
仅当潜在益处大于潜在风险时,才可在妊娠期间使用达芦那韦/利托那韦。
哺乳
还不清楚达芦那韦是否在人母乳中分泌。在大鼠中研究证明达芦那韦可在乳汁中分泌。因为在哺育婴儿,存在HIV传播和发生严重副作用的潜在风险,如果母亲正在接受达芦那韦,应告知她不要母乳喂养。
生育
在大鼠中,达芦那韦治疗对交配或生育没有影响(见毒理研究部分)。
在这些人群中,达芦那韦/利托那韦的安全性和疗效研究正在进行。目前尚无相关资料(见药代动力学部分)。
老年人:因在65岁及以上的患者中使用达芦那韦/利托那韦的资料有限,因此老年患者应慎用达芦那韦,有肝功能降低及伴随疾病或其他治疗增加的可能性(见药代动力学部分)。
达芦那韦和利托那韦都是CYP3A4异构体的抑制剂。达芦那韦和利托那韦与主要由CYP3A4代谢的药物同时使用时,可导致这些药物血浆浓度升高,继而增加或延长其疗效和副作用。
达芦那韦/利托那韦不应与高度依赖CYP3A4清除的药物同服,因它们血浆浓度的升高与严重的和/或威胁生命的事件相关(较窄的治疗指数)。这些药物包括阿司咪唑、特非那丁、咪达唑仑、三唑仑、西沙必利、哌咪清和麦角生物碱(如麦角胺、双氢麦角胺、麦角新碱和甲基麦角新碱(见禁忌部分)。
利福平是CYP450代谢的强力诱导剂。达芦那韦/利托那韦不应与利福平联合使用,因同服可引起达芦那韦血浆浓度的明显降低,导致达芦那韦疗效丧失(见注意事项部分)。
达芦那韦/利托那韦不应与含有圣约翰草(贯叶连翘)的产品伴随使用,因为同服可引起达芦那韦血浆浓度的明显降低,导致达芦那韦疗效丧失(见注意事项部分)。
抗逆转录病毒药物
核苷(酸)类逆转录酶抑制剂(N(t)RTIs)
去羟肌苷
达芦那韦/利托那韦(600/100mg b.i.d.)不会对去羟肌苷的暴露造成显著影响。达芦那韦与100mg利托那韦和去羟肌苷联合应用时,无需进行剂量调整。
去羟肌苷宜空腹服用,因此,应在达芦那韦/利托那韦(它与食物服用)1小时前或2小时后服用。
替诺福韦
与替诺福韦(替诺福韦酯300mg每天1次q.d.)相互作用的试验结果显示,当与达芦那韦/利托那韦(300/100mg b.i.d.)同服时,替诺福韦的全身暴露量增加22%,该结果在临床上意义不大。在同服期间,替诺福韦或达芦那韦的尿中排泄没有变化。替诺福韦对达芦那韦的暴露量没有明显的影响。当这些药物同服时,不需要调整达芦那韦、利托那韦或替诺福韦的剂量。
其它的NRTIs
其它的NRTIs(齐多夫定,扎西他滨,恩曲他滨,司他夫定,拉米夫定和阿巴卡韦)主要经过肾脏排泄,与达芦那韦清除途径不同,预期这些药物与达芦那韦/利托那韦之间没有相互作用。
非核苷类逆转录酶抑制剂(NNRTIs)
依曲韦林(Etravitine)
一项针对达芦那韦/利托那韦(600/100mg b.i.d.)与依曲韦林的相互作用的研究中,在达芦那韦/利托那韦存在的情况下,依曲韦林的暴露降低了37%,而达芦那韦的暴露未出现相应的改变。因此,达芦那韦/利托那韦可与依曲韦林200mg b.i.d.合用且不需要调整剂量。
依非韦伦
已经进行达芦那韦/利托那韦(300/100mg b.i.d.)与依非韦伦(600mg q.d.)之间的相互作用试验。与依非韦伦合用时,达芦那韦的暴露量降低13%。当使用达芦那韦/利托那韦时,依非韦伦的暴露量增加21%。这种差异临床上没有意义,达芦那韦/利托那韦与依非韦伦可联合使用,无需调整剂量。
奈韦拉平
达芦那韦/利托那韦(400/100mg b.i.d.)与奈韦拉平(200mg b.i.d.)相互作用的试验结果显示,与奈韦拉平同时服用时,达芦那韦暴露量不受影响。当与达芦那韦/利托那韦合用时,奈韦拉平的暴露量增加27%(比起过去的对照)。这种差异在临床上没有意义,达芦那韦/利托那韦与奈韦拉平可联合使用,无需调整剂量。
蛋白酶抑制剂(PIs)
利托那韦
当达芦那韦600mg单次剂量与利托那韦100mg b.i.d.同服时,利托那韦的总体药代动力学效应可使达芦那韦的全身暴露量提高14倍。因此,达芦那韦应以100mg利托那韦作为药代动力学增强剂与之联合使用(见注意事项和药代动力学部分)。
洛匹那韦/利托那韦
本品,加或者不加利托那韦与洛匹那韦/利托那韦(1200mg达芦那韦b.i.d.加或不加100mg利托那韦b.i.d.与洛匹那韦/利托那韦400/100mg b.i.d.或533/133.3mg b.i.d.)相互作用的试验结果显示,达芦那韦的暴露量(AUC)降低了40%。尚未确定合适的联合应用剂量。因此,不推荐本品/利托那韦与洛匹那韦/利托那韦联合应用。
沙奎那韦
在达芦那韦(400mg b.i.d.)、沙奎那韦(1000mg b.i.d.)与利托那韦(100mg b.i.d.)之间的相互作用研究中,沙奎那韦/利托那韦存在时,达芦那韦的暴露量降低26%;沙奎那韦的暴露量不受达芦那韦/利托那韦影响。不推荐沙奎那韦与伴或不伴有低剂量利托那韦的达芦那韦联合使用。
阿扎那韦
达芦那韦/利托那韦(400/100mg b.i.d.)与阿扎那韦(300mg q.d.)之间的相互作用研究显示,同服时,达芦那韦和阿扎那韦的全身暴露量没有受到显著影响。阿扎那韦可以与达芦那韦/利托那韦同服。
茚地那韦
达芦那韦/利托那韦(400/100mg b.i.d.)与茚地那韦(800mg b.i.d.)之间相互作用的研究显示,茚地那韦/利托那韦存在时,达芦那韦暴露量增加24%;达芦那韦/利托那韦存在时,茚地那韦的暴露量增加23%。当与达芦那韦/利托那韦联合使用时,在不耐受的患者中,茚地那韦的剂量从800mg b.i.d.调整到600mg b.i.d.可能是合理的。
其他蛋白酶抑制剂
达芦那韦/利托那韦与洛匹那韦/利托那韦、沙奎那韦、阿扎那韦及茚地那韦之外的PI联合使用的研究尚未开展,因此,不推荐与这些药物联合使用。
CCR5拮抗剂
当和本品/利托那韦联用时,马拉维若(Maraviro)的剂量应为150mg每日两次。在本品/利托那韦(600/100mg b.i.d.)和Maraviroc(150mg b.i.d.)的相互作用试验中,表明本品/利托那韦存在时,Maraviroc的暴露量增加305%。Maraviroc对本品/利托那韦的暴露量无明显作用。
其他药物
抗心律失常药物(苄普地尔,全身性利多卡因,奎尼丁及胺碘酮)
与达芦那韦/利托那韦同服时,苄普地尔,利多卡因,奎尼丁及胺碘酮的暴露量可能增加。需要谨慎用药,条件许可时,建议对抗心律失常药物进行监测。
地高辛
达芦那韦/利托那韦(600/100mg b.i.d.)与单次给予地高辛(0.4mg)的相互作用试验表明,地高辛AUClast增加了77%(最小二乘法均值(LSM)的比值为1.77,90%CI为0.90至3.50)。当地高辛与达芦那韦/利托那韦合用时,推荐起始采用最低剂量,然后逐渐递增剂量至获取满意的临床效果。应检测血浆地高辛浓度以帮助进行滴定。
抗凝药物
当与达芦那韦/利托那韦同服时,华法林浓度可能受到影响。华法林与达芦那韦/利托那韦联合使用时,建议监测国际标准化比值(INR)。
抗惊厥药物(苯巴比妥,苯妥类,卡马西平)
苯巴比妥和苯妥类
苯巴比妥,苯妥类是CYP450酶的诱导剂。达芦那韦/利托那韦不应与这些药物联合使用,因为同服可引起达芦那韦的血浆浓度明显降低,导致达芦那韦的疗效丧失(见注意事项部分)。
卡马西平
一项针对达芦那韦/利托那韦(600/100mg b.i.d.)与卡马西平(200mg b.i.d.)相互作用试验表明,达芦那韦与利托那韦联用时,其暴露不会受到卡马西平的影响。利托那韦的暴露(AUC12h)降低了49%。卡马西平的AUC12h增加了45%。不推荐达芦那韦/利托那韦进行剂量调整。如果需要联用达芦那韦/利托那韦和卡马西平,应监测患者是否出现了卡马西平相关的不良事件。应监测卡马西平的浓度并逐渐滴定其剂量至足量。基于该试验的结果,在于达芦那韦/利托那韦联用时,卡马西平的剂量应降低25%至50%。
钙通道阻滞剂
当与达芦那韦/利托那韦合用时,钙通道阻滞剂(如非洛地平,硝苯地平,尼卡地平)的暴露量可能增加。需要谨慎用药,推荐进行密切的临床监测。
克拉霉素
达芦那韦/利托那韦(400/100mg b.i.d.)与克拉霉素(500mg b.i.d.)之间相互作用的试验表明,克拉霉素的暴露量增加57%,而达芦那韦的暴露量不受影响。对于肾脏损害的病人,克拉霉素的剂量应减少。
地塞米松
地塞米松全身用药可诱导CYP3A4,降低达芦那韦的暴露量,导致治疗效果的丧失。因此,应谨慎联合使用。
氟替卡松丙酸酯
吸入性氟替卡松丙酸酯与达芦那韦/利托那韦合用时,可增加氟替卡松丙酸酯的血浆浓度。应选用其他药物,尤其是长期使用时。
HMG-CoA还原酶抑制剂
与达芦那韦/利托那韦同服时,高度依赖CYP3A4代谢的HMG-COA还原酶抑制剂,如洛伐他汀和辛伐他汀,预期血浆浓度会显著升高。HMG-CoA还原酶抑制剂浓度升高可引起肌病,包括横纹肌溶解。因此,不推荐达芦那韦/利托那韦与洛伐他汀及辛伐他汀合用。
与阿托伐他汀的相互作用试验结果表明,阿托伐他汀(10mg q.d.)与达芦那韦/利托那韦(300/100mg b.i.d.)联合使用时,阿托伐他汀(40mg q.d.)的暴露量比单独使用仅降低15%。当需要阿托伐他汀与达芦那韦/利托那韦合用时,推荐阿托伐他汀的开始剂量为10mg q.d.,根据临床反应逐渐增加阿托伐他汀的剂量。
达芦那韦/利托那韦(600/100mg b.i.d.)可使单次剂量普伐他汀(40mg)的暴露量增加大约80%,但又在部分受试者中发生。当需要将普伐他汀与本品/利托那韦联用时,推荐普伐他汀以可能的最小剂量开始,并逐渐滴定至满意的临床效果,同时,应监测其安全性。
H2-受体拮抗剂和质子泵抑制剂
奥美拉唑(20mg q.d.)或雷尼替丁(150mg b.i.d.)与达芦那韦/利托那韦(400/100mg b.i.d.)合用时,不影响达芦那韦的暴露量。基于这些结果,达芦那韦/利托那韦可与H2受体拮抗剂及质子泵抑制剂同服,而无需调整剂量。
免疫抑制剂(环孢霉素、他克莫司、西罗莫司)
与达芦那韦/利托那韦同服时,环孢霉素、他克莫司、西罗莫司的暴露量增加。与达芦那韦/利托那韦同服时,推荐对免疫抑制剂进行监测。
酮康唑、伊曲康唑和伏立康唑
酮康唑、伊曲康唑和伏立康唑都是CYP3A4的强力抑制剂和底物。全身使用酮康唑、伊曲康唑或伏立康唑和达芦那韦/利托那韦时,可增加达芦那韦的血浆浓度;同时,达芦那韦/利托那韦也可增加酮康唑、伊曲康唑或伏立康唑的血浆浓度。在一项相互作用试验中证实,酮康唑(200mg b.i.d.)和达芦那韦/利托那韦(400/100mg b.i.d.)同时使用时,酮康唑和达芦那韦的暴露量分别增加212%和42%。当需要同服时,酮康唑或伊曲康唑每天的剂量不应超过200mg。尽管伏立康唑的代谢也涉及CYP3A4之外的酶,当与达芦那韦/利托那韦同服时,伏立康唑的暴露量预期会增加。
美沙酮
一项研究本品/利托那韦(600/100mg b.i.d.)对稳定美沙酮维持治疗影响的相互作用试验表明,R-美沙酮AUC降低16%。根据药代动力学和临床试验结果,当美沙酮与本品/利托那韦同服时,无需进行剂量调整。然而,因为可能需要对一些患者进行维持治疗调整,因此应进行临床监测。
雌激素为基础的避孕药物
本品/利托那韦(600/100mg b.i.d.)与炔雌醇和炔诺酮的相互作用试验结果表明,稳态时对炔雌醇和炔诺酮的暴露分别降低44%和14%。可使用其他非激素类避孕药进行替代。
PDE-5抑制剂
在一项相互作用试验中,以西地那非100mg单剂作为对照,比较了西地那非25mg单剂与达芦那韦/利托那韦(400/100mg b.i.d.)合用时西地那非的全身暴露量。应谨慎同时使用PDE-5抑制剂和达芦那韦/利托那韦。如果有指征需要同时使用达芦那韦/利托那韦与西地那非、伐地那非或他达拉非时,推荐西地那非48小时内单次剂量不超过25mg,伐地那非72小时内单剂量不超过2.5mg或他达拉非72小时内单次剂量不超过10mg。
利福布丁(利福布汀)
利福布丁(利福布汀)是CYP450酶的底物。在一项有关药物相互作用的试验中,达芦那韦/利托那韦(600/100mg b.i.d.)与利福布汀(150mg隔日一次[q.o.d.])联用时,达芦那韦的全身暴露增加了57%。基于本品/利托那韦的安全性特点,达芦那韦暴露在利福布汀存在时所出现的增加并不需要对本品/利托那韦进行剂量调整。相互作用试验表明,利福布汀在以300mg q.d.单用和以150mg q.o.d.与本品/利托那韦(600/100mg b.i.d.)联用时的全身暴露是相当的,其活性代谢产物25-O去乙酰利福布汀的暴露增加。在接受二者联用的患者中,应将利福布汀的常规剂量300mg/day降低75%(即利福布汀150mg q.o.d.),同时加强对利福布汀相关不良事件的监测。
选择性5-羟色胺再摄取抑制剂(SSRIs)
帕罗西汀(20mg q.d.)或舍曲林(50mg q.d.)与达芦那韦/利托那韦(400/100mg b.i.d.)之间的相互作用试验中,帕罗西汀或舍曲林不影响达芦那韦的暴露量。达芦那韦/利托那韦存在时,舍曲林和帕罗西汀的暴露量分别降低49%和39%。如果SSRIs和达芦那韦/利托那韦同服时,应根据抗抑郁药物应答的临床评估,对SSRIs的剂量进行仔细摸索。另外,帕罗西汀或舍曲林剂量稳定的患者在使用达芦那韦/利托那韦初期,应监测抗抑郁药物的应答。
达芦那韦/利托那韦急性过量用药经验有限。达芦那韦口服液3200mg或片剂1600mg单次剂量与利托那韦联合使用时,在健康志愿者中没有发现明显不良反应。
达芦那韦过量时没有特殊的解毒药。达芦那韦过量的治疗主要是一般支持疗法,包括对患者的生命体征监测和临床状态的观察。如果有指征,可通过呕吐或胃部灌洗来清除未吸收的活性药物。也可使用活性炭去除未吸收的活性药物。由于达芦那韦与蛋白质高度结合,透析对活性药物的完全清除可能没有帮助。
本品/利托那韦疗效的证据来自对两项正在进行的随机、对照试验的24周结果分析,这两项试验的受试者均为曾经有接受抗病毒治疗经历的HIV-1成人患者(POWER1和POWER2试验)。这些结果又被两项开放标签试验TMC114-C215和TMC114-C208的24周汇总分析所证实(POWER3分析)。
POWER1和POWER2是针对高水平PI耐药患者的随机、对照、IIb期临床试验,每项试验由两部分组成:第一部分为部分设盲、剂量摸索;第二部分的期限较长,所有随机分配到本品/利托那韦治疗组的患者,推荐剂量为600/100mg b.i.d.。
这些试验中,受试者的入选条件包括,血浆HIV-1RNA>1000copies/ml,曾经接受蛋白酶抑制剂、非核苷类及核苷类逆转录酶抑制剂治疗,在筛查期至少携带一种PI主要突变,在筛查时稳定服用含有PI的方案至少8周。根据PI突变的数目、筛查时的病毒载量和是否使用恩夫韦肽进行随机分组。分析的对象包括318例POWER1试验中和319例POWER2试验中完成24周治疗或提前中止的患者。
本品/利托那韦与对照组之间人口统计学和基线特征无显著性差异。在两项试验中,共有131例患者服用本品/利托那韦600/100mg b.i.d.,平均年龄为43.0岁(范围27~73岁),其中男性89%,百人81%,黑人10%,希伯来人7%;平均基线血浆HIV-1RNA为4.61log10 copies/ml(范围是2.99~6.44log10 copies/ml);平均基线CD4细胞计数为153×106/l(范围是3~776×106/l);达芦那韦FC的平均值为4.3。在本品/利托那韦600/100mg b.i.d.治疗组,患者曾经服用的抗病毒药物中平均4种蛋白酶抑制剂、5种核苷类逆转录酶抑制剂和1种非核苷类逆转录酶抑制剂,而对照组为4种蛋白酶抑制剂、6种核苷类逆转录酶抑制剂和1种非核苷类逆转录酶抑制剂;本品/利托那韦组中20%的患者曾经使用恩夫韦肽,而对照组仅为17%。
与基线相比病毒载量下降1.0log10定义为病毒学应答。治疗组接受的药物为本品/利托那韦+优化基础药物组合(OBR),而对照组接受的药物为研究者选定的蛋白酶抑制剂+OBR,OBR由至少两种核苷类药物伴或不伴恩夫韦肽组成。在第24周时评价了两组的病毒学疗效。基于耐药试验和既往的用药史,对照组选用的蛋白酶抑制剂包括:洛匹那韦/利托那韦(36%),安普那韦或夫沙那韦(34%),沙奎那韦(35%)和阿扎那韦(17%)。
对照组中23%的患者使用增效的双蛋白酶抑制剂,所有患者中47%的患者使用恩夫韦肽,其中35%的患者是首次应用恩夫韦肽。
POWER3:其他的本品/利托那韦600/100mg b.i.d.的疗效数据是来自针对曾经接受治疗的患者的两项非随机试验TMC114-C215和TMC114-C208。试验共有246例患者纳入24周POWER3疗效分析,治疗采用本品/利托那韦,剂量为600/100mg b.i.d.,OBR由至少两种核苷类药物伴或不伴恩夫韦肽组成。入选标准与POWER1和POWER2相同,基线特征与POWER1和POWER2具有可比性。基线时血浆HIV-1RNA的中位值为4.60log10 copies/ml(范围是1.69~6.43log10 copies/ml),CD4细胞计数的中位值是115×106/l(范围是0~831×106/l),达芦那韦FC的中位值是3.2。患者曾经暴露于5种PIs、6种NRTIs和1种NNRTI,30%的患者曾经使用恩夫韦肽。基线特征基于TMC114-C215和TMC114-C208收录的327例患者,而疗效分析则基于完成24周治疗或提前退出的246例患者的中期数据。
下表显示的是POWER1和POWER2试验以及POWER3对采用品/利托那韦600/100mg b.i.d.治疗24周的疗效结果
http://x1.webres.medlive.cn/drugref/ChemPreparationDetail/201511231128480592.jpg
1)未完成治疗者退定为治疗失败:提前退出者病毒载量的变化推定为0.
2)统计处理结果基于最小二乘方,来源于含有分层因素的ANOVA模型,P值<0.001.
3)末次观察前推法
4)根据TLOVR分析法进行推算
5)奇数比来源于包含分层因素的逻辑回归模型,各分组间应答率和变化基于已观察数据,P值<0.001.
在POWER1和POWER2汇总分析中,本品/利托那韦(600/100mg b.i.d.)治疗组病毒载量对数值较基线时下降患者的比例明显超过对照组。在第24周时,病毒载量至少降低1.0log10的患者在本品/利托那韦治疗组所占的比例为70%,而对照组为21%;本品/利托那韦组中HIV-1RNA<50copies/ml的患者占45%,对照组为12%。
POWER3的24周疗效分析证实了在POWER1和POWER2试验中所见的病毒载量下降和CD4+细胞计数增加。在纳入24周分析的246例患者中,65%出现病毒学应答(血浆病毒载量较基线时至少下降1.0log10),40%病毒载量达到50copies/ml以下。
此外,本品/利托那韦600/100mg b.i.d.长期疗效的数据来源于POWER1 and POWER2 试验对经治患者48周治疗的汇总分析。对于治疗48周或提前退出的患者,48周汇总分析的结果显示,病毒学反应依然良好,在24周和48周时同样比例的患者病毒载量达到检测不到的水平(HIV RNA<50copies/ml)(分别为45%和45%)。
本品/利托那韦对经治患者的抗病毒活性
POWER1和POWER2 II期试验和POWER3分析中,458例经历广泛抗病毒治疗的患者接受本品/利托那韦治疗,采用的剂量为600/100mg b.i.d.。
本品/利托那韦治疗过程中对体内耐药病毒的选择
在POWER1和POWER2 II期试验和POWER3分析中,受试者为高度经治的患者,接受本品/利托那韦治疗。无论是病毒载量反弹(50例),还是病毒载量从未得到有效抑制(70例),均为病毒学失败的患者,从这些患者中分离的蛋白酶多重耐药HIV-1病毒株的分析显示,这些病毒株出现与达芦那韦敏感性降低有关的氨基酸替换。在经本品/利托那韦治疗病毒学失败的患者标本中分离的病毒株中,氨基酸替换大于20%的位点为V32I和I54L,在10~20%之间者为L33F、I47V和L89V。
与其他蛋白酶之间的体内交叉耐药
目前关于本品/利托那韦选择的病毒株的交叉耐药情况所知甚少。对于本品/利托那韦100/600mg b.i.d.治疗组病毒反跳患者中分离的病毒株,与基线相比,终点时本品FC却几乎没有增加(平均增加0.82),提示这两种蛋白酶抑制剂之间交叉耐药的程度有限。基线时对替拉那韦耐药的患者(FC>3),在24周时,病毒载量的变化的平均值为-1.38log10。由于在基线时病毒已经对其他蛋白酶抑制剂出现耐药,因而无法研究这些药物的FC增加。对于基线时无敏感蛋白酶抑制剂可用的患者(不包括替拉那韦),24周时病毒载量变化的平均值为-1.57log10。
基线时基因型耐药或表型耐药及病毒学检测结果
通过分析数据,评价了基线存在的一些特定蛋白酶抑制剂(PI)耐药突变对病毒学疗效的影响。在POWER1和POWER2试验中,基线时存在的V32I、I47V或I54L/M突变与达芦那韦的病毒学疗效下降和敏感性降低有关。此外,基线时存在7个以上PI耐药相关突变(在30、32、36、46、47、48、50、53、73、82、84、88或90位点出现任何改变)的患者病毒学疗效极差。然而,与对照组相比,达芦那韦/利托那韦治疗的各个亚组(基线时根据突变的类型和数量分组)发生病毒学应答的比例通常较高。
在POWER1和POWER2辅助分析和POWER3分析中,基线时存在以下3种或以上突变与PREZISTA/rtv病毒学疗效降低有关,这些突变包括V11I、V32I、L33F、I47V、I50V、I54L/M、G73S、L76V、I84V和L89V。
根据基线时基因型*,本品/rtv600/100mg b.i.d.的病毒学疗效:POWER1、POWER2和POWER3处理分析的结果
http://x1.webres.medlive.cn/drugref/ChemPreparationDetail/201511231131490163.jpg
*与本品/rtv病毒学疗效极差有关的突变的数量(V11I、V32I、L33F、I47V、I50V、I54L/M、G73S、L76V、I84V和L89V)
特定突变或突变类型与疗效相关性的结论可能会随着进一步研究的数据而发生变化。
基线时达芦那韦的表现型(相对于参照而发生的敏感性变化)是病毒学疗效的预测因素。
根据基线时达芦那韦表现型而评价的病毒学应答比例见下表。这些基线时表型分组基于POWER1、POWER2试验和POWER3分析入选的病人人群,并不代表全面的临床敏感性数据。基于PI经治患者治疗前对达芦那韦的敏感性,这些数据向临床医师提供了病毒学成功可能性的信息。
根据基线时表型,本品/rtv 600/100mg b.i.d.的病毒学疗效:POWER1、POWER2和POWER3处理分析的结果
对于曾经抗病毒治疗失败的患者,在选择新的治疗方案时,在可能的情况下,应当慎重考虑治疗史以及耐药检测的结果。
药理作用
作用机理
达芦那韦是一种HIV-1蛋白酶抑制剂,选择性抑制病毒感染细胞中HIV编码的Gag-Pol多蛋白的裂解,从而阻止成熟的感染性病毒颗粒的形成。
达芦那韦与HIV-1蛋白酶紧密结合,Kp值为4.5×10-12M。达芦那韦对于蛋白酶抑制剂耐药相关的突变(RAM)具有一定的疗效,但达芦那韦对目前检测到的13种人体细胞蛋白酶没有抑制作用。
体外抗逆转录病毒活性
在急性感染的T细胞系,人外周血单核细胞和人单核/巨噬细胞中,达芦那韦具有抗HIV-1实验室株和临床分离株以及HIV-2实验室株和临床分离株的活性,EC50的范围在1.2~8.5nM(0.7~5.0ng/ml)。达芦那韦在体外具有广谱抗HIV-1活性,对于M组(A、B、C、D、E、F、G亚型)和O组原始分离株均具有抗逆转录病毒活性,EC50的范围在<0.1~4.3nM,这些值显著小于50%细胞毒浓度范围(87μM~>100μM)。
在人血清中,达芦那韦的EC50增加的中位因子为5.4。达芦那韦与蛋白酶抑制剂利托那韦、奈非那韦、或安普那韦联合使用时具有协同作用,与蛋白酶抑制剂茚地那韦、沙奎那韦、洛匹那韦、阿扎那韦、或替拉那韦,核苷(酸)类逆转录抑制剂齐多夫定、拉米夫定、扎西他滨、去羟肌苷、司他夫定、阿巴卡韦、恩曲他滨或替诺福韦,非核苷类逆转录酶抑制剂etravirine、奈韦拉平、地拉韦啶或依非韦伦,以及融合抑制剂恩夫韦肽联合使用时具有相加作用。没有发现达芦那韦与其他抗逆转录病毒药物之间具有拮抗作用。
体外耐药
HIV-1体外试验中,从野生株中选择出达芦那韦耐药株是一个漫长的过程(长于2年),在达芦那韦浓度大于200nM时,选择的病毒株不能生长。这些选择性病毒株对达芦那韦的敏感性降低(范围6~21倍),蛋白酶序列发生3~6个氨基酸替换,目前正在鉴定和研究对达芦那韦敏感性降低的耐药决定簇。
对于携有多PI耐药相关突变的9个HIV病毒株,体外药物选择试验显示,达芦那韦耐药HIV-1(EC50的变化范围:53~641倍)在蛋白酶序列出现22处突变,9例达芦那韦耐药分离株中L10F、V32I、L33F、S37N、M46I、I47V、I50V、L63P、A71V和I84V出现的频率在50%以上。对达芦那韦耐药(倍数变化[FC]>10)的HIV-1株,其蛋白酶序列上至少需要8个达芦那韦体外选择的突变,其中在选择前蛋白酶序列上已存在至少2个突变。
在对安普那韦、阿扎那韦、茚地那韦、洛匹那韦、奈非那韦、利托那韦、沙奎那韦和/或替拉那韦耐药的1113例临床分离株,以及POWER1、POWER2和POWER3试验收录的886例基线分离株中,只有携带10个以上PI耐药相关突变的病毒株对达芦那韦耐药的平均倍数变化(FC)才大于10。
体外交叉耐药
蛋白酶抑制剂之间存在交叉耐药。对安普那韦、阿扎那韦、茚地那韦、洛匹那韦、奈非那韦、利托那韦、沙奎那韦和/或替拉那韦耐药的3309例临床分离株中,90%对达芦那韦的敏感性下降<10倍,这说明对大多数PI耐药的病毒株对达芦那韦仍然敏感。
从PI耐药病毒株中选择的9例对达芦那韦耐药的病毒株中7例具有替拉那韦的表型耐药试验资料,其中6例替拉那韦EC50的倍比变化小于3,提示这两种蛋白酶抑制剂之间交叉耐药的程度有限。
达芦那韦与核苷(酸)类逆转录酶抑制剂、非核苷酸类逆转录酶抑制剂、侵入抑制剂或整合酶抑制剂之间不可能存在交叉耐药,因为达芦那韦与这些抑制剂之间作用于病毒的靶位不同。
毒理研究
达芦那韦单药的动物毒性试验已在小鼠、大鼠和犬中进行,与利托那韦合用的毒性试验已在大鼠和犬种进行。
在大鼠和犬慢性毒性试验中,达芦那韦仅有局限的毒副作用。在大鼠试验中,暴露量在100mg/kg/天或以上和低于临床暴露水平时,药物作用的主要靶器官为造血系统、凝血系统、肝脏和甲状腺。许多红细胞相关的参数出现小幅下降,伴有激活的PTT(部分促凝血酶原激酶时间)延长。肝脏和甲状腺的变化可能是对大鼠酶诱导作用的适应性反应,而非不良反应。在与利托那韦合用药的毒性试验中,在大鼠中没有关于其它靶器官毒性的报道。高达120mg/kg/天的剂量以及在临床推荐剂量的暴露量之下,在犬种没有发现严重的毒性反应或靶器官损害。
大鼠试验显示,达芦那韦剂量达1000mg/kg/天时对交配和繁殖没有影响,暴露水平(AUC-0.5 fold)低于人体临床推荐的治疗量。在同样剂量水平下,达芦那韦单药对大鼠和兔没有致畸作用,与利托那韦合用时对小鼠也没有致畸作用,暴露水平低于人体临床推荐的剂量。在大鼠出生前后的发育评估试验中,达芦那韦无论是否与利托那韦联合均可引起哺乳期子鼠体重的短期下降,这可能与通过乳汁导致的药物暴露有关,在断奶后,药物对子鼠的各项功能没有影响。
在直接接受达芦那韦(从20mg/kg至1000mg/kg)的年龄在23天至25天的幼年大鼠中观察到了死亡的现象,同时,在一些动物中,还观察到惊厥的现象。在这个年龄范围内,血浆、肝脏和大脑中的暴露量取决于剂量和年龄,同时,该暴露量大大高于在成年大鼠中观察到的暴露量。这些结果的出现与达芦那韦代谢涉及的CYP450肝酶的个体发生与脑血屏障不成熟有关。年龄为26天的幼年大鼠接受1000mg/kg达芦那韦(单次给药)或年龄为23至50天的幼年大鼠接受500mg/kg(重复给药)时,未观察到治疗相关死亡,同时,其暴露和毒性特点与成年大鼠中所观察到的特点相当。在人体中,药物代谢酶的活性在3岁时即已达成年时的数值。
通过管饲法给药(最多104周)评价了达芦那韦在小鼠和大鼠中的致癌性。小鼠接受的每日剂量包括150、450和1000mg/kg,而大鼠接受的每日剂量包括50、150和500mg/kg。在两个物种的雌性和雄性中均观察到肝细胞腺瘤和肝细胞癌的发生率随剂量的增加而增加。在雄性大鼠中观察到了甲状腺滤泡细胞腺瘤。达芦那韦给予小鼠或大鼠后并未导致其他任何良性或恶性肿瘤的发生率出现具有显著统计学意义的增加。所观察到的在啮齿类动物中的肝细胞结果被认为与人类的相关性有限。重复给予大鼠达芦那韦可诱导肝微粒体酶并增加甲状腺激素的清除,这些会导致大鼠(但非人类)更易发生甲状腺肿瘤。在所测试的最高剂量下,达芦那韦的系统暴露(基于AUC)分别是人接受推荐治疗剂量(600/100mg、每天两次或800/100mg、每天一次)时所观察到的暴露的0.4至0.7倍(小鼠)和0.7至1倍(大鼠)。达芦那韦在细菌回复突变试验(Ames)、在人淋巴细胞内的染色体畸变以及在小鼠体内的微核试验中,结果均为阴性。
本品与利托那韦合用的药代动力学特点,在健康成人志愿者和HIV-1感染者中已获得评估。HIV-1感染者达芦那韦的暴露量高于健康受试者,其原因可能在于HIV-1感染者α-1-酸性糖蛋白(AAG)的浓度较高,达芦那韦与血浆AAG结合较多,从而导致达芦那韦血浆浓度较高。
达芦那韦主要通过CYP3A4代谢,利托那韦可抑制CYP3A4的活性,因此,在与利托那韦合用时,达芦那韦的血浆浓度会显著增加。
吸收
口服后,达芦那韦快速吸收。与低剂量利托那韦同服时,达芦那韦的最大血浆浓度通常在服药后2.5~4.0小时达到。
600mg单剂量本品的绝对口服生物利用度大约为37%,与利托那韦100mg b.i.d.合用时,生物利用度增加到82%。在利托那韦的作用下,达芦那韦的全身暴露量增加约14倍。
与进餐时服用相比,不与食物同服时本品与低剂量利托那韦合用的相对生物利用度降低30%,因此,本品片剂应当与利托那韦和食物同服,食物的类型不影响达芦那韦的暴露量。
分布
约95%的达芦那韦与血浆蛋白结合,主要与血浆α-1-酸性糖蛋白结合。
代谢
人肝微粒体(HLM)的体外试验显示,达芦那韦主要进行氧化代谢,被肝脏CYP系统广泛代谢,绝大多数被CYP3A4同工酶代谢。一项在健康志愿者中进行的14C-达芦那韦试验显示,单次服用本品/利托那韦400/100mg后血浆中大部分放射活性物质来源于达芦那韦本身,在人体内已发现至少3种氧化代谢产物,这些产物对于野生型HIV的活性小于达芦那韦活性的至少10倍。
清除
单次服用400/100mg 14C-达芦那韦/利托那韦后,粪便和尿液中检测的14C-达芦那韦大约分别占79.5%和13.9%。达芦那韦原药大约分别占粪便和尿液中剂量的41.2%和7.7%。与利托那韦合用时,达芦那韦的终末清除半衰期约为15小时。达芦那韦(150mg)单独用药以及与低剂量利托那韦合用是,静脉清除率分别为32.8l/h和5.9l/h。
特殊人群
儿童
本品与利托那韦合用在儿童受试者中的药代动力学尚在研究阶段,目前没有关于儿童的推荐剂量。
老年人
人群药代动力学分析显示,HIV感染人群(n=12,年龄≥65岁)本品的药代动力学在18~75岁这一年龄范围内并无明显差异(见注意事项部分)。
性别
人群药代动力学分析显示,女性达芦那韦的暴露量略高于男性(16.8%),这一差异无临床意义。
肾功能障碍
14C-达芦那韦/利托那韦的质量守恒试验的结果显示,大约7.7%的达芦那韦以原型从尿中排出。
尽管本品没有在肾功能障碍的患者中进行研究,人群药代动力学分析显示,本品的药代动力学在中度肾功能障碍(肌酐清除率在30~60ml/分钟,n=20)的HIV患者中没有显著影响(见用法用量及注意事项部分)。
肝功能障碍
在一项针对本品/利托那韦(600/100mg)、每日两次的多次给药研究结果表明,达芦那韦在轻度(Child Pugh A级,n=8)和中度(Child Pugh B级,n=8)肝损伤受试者体内的稳态药代动力学参数与其在健康受试者的参数相当。尚未研究严重肝功能损伤对达芦那韦的药代动力学影响(见用法用量及注意事项部分)。
低于30℃保存。
高密度聚乙烯塑料瓶装,配有儿童不易打开的瓶盖。120片/瓶/盒。
24个月
JX20070057
成份
主要成份:达芦那韦
性状
本品为白色薄膜衣异形片,除去包衣后显白色或类白色。
适应症
达芦那韦联合100mg利托那韦合用(达芦那韦/利托那韦),和其他抗逆转录病毒药物合并使用,适用于已使用过抗逆转录病毒药物的HIV(人类免疫缺陷病毒)感染的成人患者的治疗,例如对一种以上蛋白酶抑制剂耐药的HIV-1感染者。
这一适应症基于两项达芦那韦/利托那韦与其他抗病毒药物联合的对照试验中血浆HIV RNA水平与CD4+细胞计数24周分析结果。两项试验的受试者均为临床晚期、经历抗病毒治疗(NRTIs、NNRTIs和Pis)的成人患者,这些患者尽管在进行抗病毒治疗但未能抑制HIV-1复制。
对于初治患者或儿童患者,达芦那韦/利托那韦的风险和收益缺乏相关资料。
规格
300mg。
用法用量
使用方法:口服。
使用达芦那韦时,必须以100mg利托那韦作为药代动力学增效剂,并联合其它抗逆转录病毒药物。因此在开始达芦那韦/利托那韦治疗前,必须参考利托那韦的处方资料。
成人:达芦那韦的推荐剂量是每次60mg,每天2次(b.i.d.),与利托那韦(每次100mg,每天2次)及食物同服。食物的类型不影响达芦那韦的吸收。利托那韦(每次100mg,每天2次)用来作为达芦那韦的药代动力学增效剂(见药物相互作用和药代动力学部分)。进一步增加达芦那韦或利托那韦的剂量不太可能引起临床抗病毒活性的相应增加。
儿童患者:达芦那韦在儿童患者中的安全性和有效性尚未通过验证。
肝脏损害:对于肝功能损害的患者,目前尚无使用达芦那韦/利托那韦的相关资料,因此没有具体的推荐剂量。肝脏损害的患者应慎用达芦那韦/利托那韦(见注意事项及药代动力学部分)。
肾脏损害:对于中度肾功能障碍的患者,无需调整剂量,对于重度或终末期肾病的HIV-1感染患者,尚缺乏药代动力学数据(见注意事项及药代动力学部分)。
不良反应
1.1在汇总的POWER 1、2和3中发现的本品/利托那韦的药物不良反应
在汇总的POWER试验中,立即开始使用本品/利托那韦600/100mg、b.i.d.治疗的患者的总暴露为812.4患者年。
在本品/利托那韦治疗期间所报告的大部分不良反应(ADR)均为轻度。
最常见的(≥5%)中重度(2-4级)ADR包括腹泻、头痛、腹痛、恶心和呕吐。
最常见的3级或4级ADR包括肝酶及胰酶升高、高甘油三酯血征、腹泻、高胆固醇血征、头痛、腹痛和呕吐。所有其他3级或4级ADR的发生率均低于1%。
2.1%的患者因ADR而终止了治疗。
下表总结了在汇总的POWER1、2和3试验中本品/利托那韦600/100mg b.i.d.用于未接受过抗逆转录病毒治疗的成年HIV-1患者时发生的至少为中度(2-4级)的药物不良反应*:
http://x1.webres.medlive.cn/drugref/ChemPreparationDetail/201511231118170424.jpg
下表总结了在汇总的POWER1、2和3试验中本品/利托那韦用于接受过抗逆转录病毒治疗的成年HIV-1患者中发生的被认为是ADR的实验检查结果异常:
http://x1.webres.medlive.cn/drugref/ChemPreparationDetail/201511231119590886.jpg
1.2在其他临床试验中发现的本品/利托那韦的其他药品不良反应:Artemis[TMC114-C211],Titan[TMC114-C214],TMC114-C208,TMC114-C209,DUET_1[TMC125-C206],DUET_2[TMC125-C216]或其他正在进行中的试验。
http://x1.webres.medlive.cn/drugref/ChemPreparationDetail/201511231120500069.jpg
在上述试验或其他正在进行的试验中,未发现其他ADR术语。
2 联合抗逆转录病毒治疗的效果
HIV患者中联合抗病毒治疗与身体脂肪重新分布(脂肪代谢障碍)相关,这些异常包括外周和面部皮下脂肪丢失,腹腔和内脏脂肪增多,乳房肥大,以及颈背部脂肪堆积(水牛背)。
联合抗病毒治疗与代谢异常相关,如高甘油三酯血征,高胆固醇血征,胰岛素抵抗,高血糖征及高乳糖血征。
在开始联合抗逆转录病毒治疗时存在严重免疫缺陷的HIV感染患者,可能会出现对无症状或残留的机会性感染的炎症反应。
接受蛋白酶抑制剂(PIs)治疗血友病患者中已有自发性出血增多的报告。
使用蛋白酶抑制剂,特别是与NRTIs联合使用时,已有CPK升高,肌痛,肌炎的报道,极少数患者可出现横纹肌溶解。
3特殊人群
乙型肝炎和/或丙型肝炎共感染的患者
在乙型肝炎或丙型肝炎共感染并接受本品/利托那韦的患者中,除肝酶升高外,不良反应和临床生化检查异常的发生率不高于无共同感染并接受本品/利托那韦治疗的患者。共感染患者与无共感染患者的药代动力学暴露量相当。对肝炎共感染患者给予标准的临床监测是可行的。
禁忌
对达芦那韦或达芦那韦所含任何一种成份过敏者。
达芦那韦和利托那韦都是细胞色素P450 3A(CYP3A4)异构体的抑制剂。达芦那韦/利托那韦不应与高度依赖CYP3A4清除的药物同时服用,因为这些药物血浆浓度增高与严重和/或危及生命的事件相关(治疗指数狭窄)。这些药物包括阿司咪唑(息斯敏)、特非那丁、咪达唑仑、三唑仑、西沙比利、哌迷清和麦角生物碱(如麦角胺、双氢麦角胺、麦角新碱和甲基麦角新碱)(见药物相互作用部分)。
注意事项
1.应告知患者目前的抗逆转录病毒治疗还不能治愈HIV,而且还没有证实能够预防HIV的传播。应继续使用适当的预防措施。
2.在考虑到对幼鼠给药(20mg/kg到1,000mg/kg)23到26天观察到毒性,本品/利托那韦不能用于3岁以下儿童,(见药理毒理部分)
本品/利托那韦的安全性和有效性尚未在对已接受过抗逆转录病毒治疗的3-6岁儿童和未接受过抗逆转录病毒治疗的儿童患者中确定。
3.老年人:因在65岁及以上的患者中使用达芦那韦的资料有限,因此老年患者应慎用达芦那韦,有肝功能降低及伴随疾病或其它治疗增加的可能性(见药代动力学部分)。
4.单次剂量600mg本品的绝对口服生物利用度大约是37%,与100mg利托那韦b.i.d.合用时,增加到约82%,利托那韦的药代动力学增强效果大约可使达芦那韦的全身暴露量增加14倍。因此,本品应与低剂量的利托那韦联合使用(见药代动力学部分)。
增加利托那韦的剂量对达芦那韦的浓度无明显影响,因此不推荐增加剂量。
5.在临床试验中(n=3063),皮疹(所有级别,无论原因为何)在接受PREZISTA治疗的患者中的发生率为10.3%(见不良反应部分)。大部分皮疹均为轻至中度,常常发生在治疗的前四周内,在不停药的情况下都可以治愈。在接受本品/利托那韦治疗的患者中,因皮疹而 停药的患者占0.5%。
严重的皮疹(可能伴有发热和/或转氨酶升高)在患者中的发生率为0.4%。多形性渗出性红斑罕见(<0.1%)。如果出现了严重的皮疹,应停用本品。
6.达芦那韦含有磺胺。在已知对磺胺过敏的患者中,应慎用本品。在一项针对本品/利托那韦的临床研究中,有磺胺过敏史的患者中皮疹的发生率和严重程度与无磺胺过敏史的患者是相当的。
7.请置于儿童不易拿到处
伴随疾病的患者
肝脏损伤
目前尚无严重肝损伤患者使用本品/利托那韦的相关资料;因此,不推荐具体剂量。严重肝损伤患者应慎用本品/利托那韦。试验数据证实,轻度或中度肝损伤受试者使用达芦那韦时,其稳态药代动力学参数与健康受试者的参数接近,轻度或中度肝损伤患者无需进行剂量调整(见用法用量和药代动力学部分)。
曾患肝功能障碍的病人,包括慢性活动性乙型或丙型肝炎,在联合抗逆转录病毒治疗中肝脏功能异常的发生频率可能增加,应根据标准程序进行监测。在这些病人中,如果有肝脏疾病恶化的证据,需要考虑中断或中止治疗。
肾脏损伤
因为达芦那韦的肾脏清除有限,在肾脏损害的病人中,机体总的清除率预期不会降低。达芦那韦和利托那韦与血浆蛋白高度结合,因此它们不太可能通过血液透析或腹膜透析而大量清除(见用法用量和药代动力学部分)。
血友病患者
在使用PIs治疗的A型和B型血友病患者中,已有出血增加的报道,包括自发的皮肤血肿和关节血肿。在部分病人中给予了额外的VIII因子。半数以上的报告病例中,如果已经停止治疗,可继续或再引入PIs治疗。尽管这种导致出血的作用机制还没有被阐明,但已提示因果关系。因此,应使血友病患者意识到出血增加的可能性。
高血糖征
在接受抗逆转录病毒治疗(包括PIs治疗)的病人中,已经有新发糖尿病,高糖血征或糖尿病恶化的报道。部分病人中,高糖血征很严重,部分病例尚会出现酮症酸中毒。许多病人的病情较为复杂,其中一些情况需要药物治疗,但这些药物又可引起糖尿病或高血糖征进一步恶化。
脂肪重新分布&代谢紊乱
在HIV感染病人中,联合抗逆转录病毒治疗可引起机体脂肪的重新分布(脂肪代谢障碍)。这些副作用的长期后果目前尚不明确,对其机制的了解还不完善。目前认为内脏脂肪过多与PIs、皮下脂肪萎缩和NRTIs有一定关系,脂肪代谢障碍的高危因素包括个体因素如年龄较大,药物相关因素如抗病毒治疗持续时间较长,以及与治疗有关的代谢障碍。临床上应对脂肪重新分布的体征进行评估,检测空腹血脂和血糖,对脂质紊乱给予适当的处理(见不良反应)。
免疫重建炎性综合征
严重免疫缺陷的艾滋病患者在开始联合抗病毒治疗(CART)后,可能出现对无症状或残余的机会性病原菌的炎性反应,并引起严重的临床疾病,或症状好恶化。一般在开始CART的前几周或数月内可以观察到这种反应。有关的例子如巨细胞病毒性视网膜炎、播散性和/或局灶性分枝杆菌感染、及肺孢子虫性肺炎。对任何炎性症状应进行评估,必要时给予治疗。
药物相互作用
达芦那韦和利托那韦都是CYP3A4异构体的抑制剂。达芦那韦和利托那韦与主要由CYP3A4代谢的药物同时服用时,可导致这些药物血浆浓度增加,从而增加或延长它们的疗效和副作用(见禁忌和药物相互作用部分)。
达芦那韦经CYP3A4代谢。能够诱导CYP3A4活性的药品可能会增加达芦那韦的清除率,从而降低达芦那韦的血浆浓度(见药物相互作用部分)。
达芦那韦与其他能够抑制CYP3A4活性的药品合用时,可能会降低达芦那韦的清除率,从而增加达芦那韦的血浆浓度(见药物相互作用部分)。
孕妇及哺乳期妇女用药
妊娠
达芦那韦在妊娠妇女中的研究较少,也没有较好的对照研究。动物试验中没有发现发育毒性或影响生殖和生育功能的证据(见毒理研究部分)。
仅当潜在益处大于潜在风险时,才可在妊娠期间使用达芦那韦/利托那韦。
哺乳
还不清楚达芦那韦是否在人母乳中分泌。在大鼠中研究证明达芦那韦可在乳汁中分泌。因为在哺育婴儿,存在HIV传播和发生严重副作用的潜在风险,如果母亲正在接受达芦那韦,应告知她不要母乳喂养。
生育
在大鼠中,达芦那韦治疗对交配或生育没有影响(见毒理研究部分)。
儿童用药
在这些人群中,达芦那韦/利托那韦的安全性和疗效研究正在进行。目前尚无相关资料(见药代动力学部分)。
老年用药
老年人:因在65岁及以上的患者中使用达芦那韦/利托那韦的资料有限,因此老年患者应慎用达芦那韦,有肝功能降低及伴随疾病或其他治疗增加的可能性(见药代动力学部分)。
药物相互作用
达芦那韦和利托那韦都是CYP3A4异构体的抑制剂。达芦那韦和利托那韦与主要由CYP3A4代谢的药物同时使用时,可导致这些药物血浆浓度升高,继而增加或延长其疗效和副作用。
达芦那韦/利托那韦不应与高度依赖CYP3A4清除的药物同服,因它们血浆浓度的升高与严重的和/或威胁生命的事件相关(较窄的治疗指数)。这些药物包括阿司咪唑、特非那丁、咪达唑仑、三唑仑、西沙必利、哌咪清和麦角生物碱(如麦角胺、双氢麦角胺、麦角新碱和甲基麦角新碱(见禁忌部分)。
利福平是CYP450代谢的强力诱导剂。达芦那韦/利托那韦不应与利福平联合使用,因同服可引起达芦那韦血浆浓度的明显降低,导致达芦那韦疗效丧失(见注意事项部分)。
达芦那韦/利托那韦不应与含有圣约翰草(贯叶连翘)的产品伴随使用,因为同服可引起达芦那韦血浆浓度的明显降低,导致达芦那韦疗效丧失(见注意事项部分)。
抗逆转录病毒药物
核苷(酸)类逆转录酶抑制剂(N(t)RTIs)
去羟肌苷
达芦那韦/利托那韦(600/100mg b.i.d.)不会对去羟肌苷的暴露造成显著影响。达芦那韦与100mg利托那韦和去羟肌苷联合应用时,无需进行剂量调整。
去羟肌苷宜空腹服用,因此,应在达芦那韦/利托那韦(它与食物服用)1小时前或2小时后服用。
替诺福韦
与替诺福韦(替诺福韦酯300mg每天1次q.d.)相互作用的试验结果显示,当与达芦那韦/利托那韦(300/100mg b.i.d.)同服时,替诺福韦的全身暴露量增加22%,该结果在临床上意义不大。在同服期间,替诺福韦或达芦那韦的尿中排泄没有变化。替诺福韦对达芦那韦的暴露量没有明显的影响。当这些药物同服时,不需要调整达芦那韦、利托那韦或替诺福韦的剂量。
其它的NRTIs
其它的NRTIs(齐多夫定,扎西他滨,恩曲他滨,司他夫定,拉米夫定和阿巴卡韦)主要经过肾脏排泄,与达芦那韦清除途径不同,预期这些药物与达芦那韦/利托那韦之间没有相互作用。
非核苷类逆转录酶抑制剂(NNRTIs)
依曲韦林(Etravitine)
一项针对达芦那韦/利托那韦(600/100mg b.i.d.)与依曲韦林的相互作用的研究中,在达芦那韦/利托那韦存在的情况下,依曲韦林的暴露降低了37%,而达芦那韦的暴露未出现相应的改变。因此,达芦那韦/利托那韦可与依曲韦林200mg b.i.d.合用且不需要调整剂量。
依非韦伦
已经进行达芦那韦/利托那韦(300/100mg b.i.d.)与依非韦伦(600mg q.d.)之间的相互作用试验。与依非韦伦合用时,达芦那韦的暴露量降低13%。当使用达芦那韦/利托那韦时,依非韦伦的暴露量增加21%。这种差异临床上没有意义,达芦那韦/利托那韦与依非韦伦可联合使用,无需调整剂量。
奈韦拉平
达芦那韦/利托那韦(400/100mg b.i.d.)与奈韦拉平(200mg b.i.d.)相互作用的试验结果显示,与奈韦拉平同时服用时,达芦那韦暴露量不受影响。当与达芦那韦/利托那韦合用时,奈韦拉平的暴露量增加27%(比起过去的对照)。这种差异在临床上没有意义,达芦那韦/利托那韦与奈韦拉平可联合使用,无需调整剂量。
蛋白酶抑制剂(PIs)
利托那韦
当达芦那韦600mg单次剂量与利托那韦100mg b.i.d.同服时,利托那韦的总体药代动力学效应可使达芦那韦的全身暴露量提高14倍。因此,达芦那韦应以100mg利托那韦作为药代动力学增强剂与之联合使用(见注意事项和药代动力学部分)。
洛匹那韦/利托那韦
本品,加或者不加利托那韦与洛匹那韦/利托那韦(1200mg达芦那韦b.i.d.加或不加100mg利托那韦b.i.d.与洛匹那韦/利托那韦400/100mg b.i.d.或533/133.3mg b.i.d.)相互作用的试验结果显示,达芦那韦的暴露量(AUC)降低了40%。尚未确定合适的联合应用剂量。因此,不推荐本品/利托那韦与洛匹那韦/利托那韦联合应用。
沙奎那韦
在达芦那韦(400mg b.i.d.)、沙奎那韦(1000mg b.i.d.)与利托那韦(100mg b.i.d.)之间的相互作用研究中,沙奎那韦/利托那韦存在时,达芦那韦的暴露量降低26%;沙奎那韦的暴露量不受达芦那韦/利托那韦影响。不推荐沙奎那韦与伴或不伴有低剂量利托那韦的达芦那韦联合使用。
阿扎那韦
达芦那韦/利托那韦(400/100mg b.i.d.)与阿扎那韦(300mg q.d.)之间的相互作用研究显示,同服时,达芦那韦和阿扎那韦的全身暴露量没有受到显著影响。阿扎那韦可以与达芦那韦/利托那韦同服。
茚地那韦
达芦那韦/利托那韦(400/100mg b.i.d.)与茚地那韦(800mg b.i.d.)之间相互作用的研究显示,茚地那韦/利托那韦存在时,达芦那韦暴露量增加24%;达芦那韦/利托那韦存在时,茚地那韦的暴露量增加23%。当与达芦那韦/利托那韦联合使用时,在不耐受的患者中,茚地那韦的剂量从800mg b.i.d.调整到600mg b.i.d.可能是合理的。
其他蛋白酶抑制剂
达芦那韦/利托那韦与洛匹那韦/利托那韦、沙奎那韦、阿扎那韦及茚地那韦之外的PI联合使用的研究尚未开展,因此,不推荐与这些药物联合使用。
CCR5拮抗剂
当和本品/利托那韦联用时,马拉维若(Maraviro)的剂量应为150mg每日两次。在本品/利托那韦(600/100mg b.i.d.)和Maraviroc(150mg b.i.d.)的相互作用试验中,表明本品/利托那韦存在时,Maraviroc的暴露量增加305%。Maraviroc对本品/利托那韦的暴露量无明显作用。
其他药物
抗心律失常药物(苄普地尔,全身性利多卡因,奎尼丁及胺碘酮)
与达芦那韦/利托那韦同服时,苄普地尔,利多卡因,奎尼丁及胺碘酮的暴露量可能增加。需要谨慎用药,条件许可时,建议对抗心律失常药物进行监测。
地高辛
达芦那韦/利托那韦(600/100mg b.i.d.)与单次给予地高辛(0.4mg)的相互作用试验表明,地高辛AUClast增加了77%(最小二乘法均值(LSM)的比值为1.77,90%CI为0.90至3.50)。当地高辛与达芦那韦/利托那韦合用时,推荐起始采用最低剂量,然后逐渐递增剂量至获取满意的临床效果。应检测血浆地高辛浓度以帮助进行滴定。
抗凝药物
当与达芦那韦/利托那韦同服时,华法林浓度可能受到影响。华法林与达芦那韦/利托那韦联合使用时,建议监测国际标准化比值(INR)。
抗惊厥药物(苯巴比妥,苯妥类,卡马西平)
苯巴比妥和苯妥类
苯巴比妥,苯妥类是CYP450酶的诱导剂。达芦那韦/利托那韦不应与这些药物联合使用,因为同服可引起达芦那韦的血浆浓度明显降低,导致达芦那韦的疗效丧失(见注意事项部分)。
卡马西平
一项针对达芦那韦/利托那韦(600/100mg b.i.d.)与卡马西平(200mg b.i.d.)相互作用试验表明,达芦那韦与利托那韦联用时,其暴露不会受到卡马西平的影响。利托那韦的暴露(AUC12h)降低了49%。卡马西平的AUC12h增加了45%。不推荐达芦那韦/利托那韦进行剂量调整。如果需要联用达芦那韦/利托那韦和卡马西平,应监测患者是否出现了卡马西平相关的不良事件。应监测卡马西平的浓度并逐渐滴定其剂量至足量。基于该试验的结果,在于达芦那韦/利托那韦联用时,卡马西平的剂量应降低25%至50%。
钙通道阻滞剂
当与达芦那韦/利托那韦合用时,钙通道阻滞剂(如非洛地平,硝苯地平,尼卡地平)的暴露量可能增加。需要谨慎用药,推荐进行密切的临床监测。
克拉霉素
达芦那韦/利托那韦(400/100mg b.i.d.)与克拉霉素(500mg b.i.d.)之间相互作用的试验表明,克拉霉素的暴露量增加57%,而达芦那韦的暴露量不受影响。对于肾脏损害的病人,克拉霉素的剂量应减少。
地塞米松
地塞米松全身用药可诱导CYP3A4,降低达芦那韦的暴露量,导致治疗效果的丧失。因此,应谨慎联合使用。
氟替卡松丙酸酯
吸入性氟替卡松丙酸酯与达芦那韦/利托那韦合用时,可增加氟替卡松丙酸酯的血浆浓度。应选用其他药物,尤其是长期使用时。
HMG-CoA还原酶抑制剂
与达芦那韦/利托那韦同服时,高度依赖CYP3A4代谢的HMG-COA还原酶抑制剂,如洛伐他汀和辛伐他汀,预期血浆浓度会显著升高。HMG-CoA还原酶抑制剂浓度升高可引起肌病,包括横纹肌溶解。因此,不推荐达芦那韦/利托那韦与洛伐他汀及辛伐他汀合用。
与阿托伐他汀的相互作用试验结果表明,阿托伐他汀(10mg q.d.)与达芦那韦/利托那韦(300/100mg b.i.d.)联合使用时,阿托伐他汀(40mg q.d.)的暴露量比单独使用仅降低15%。当需要阿托伐他汀与达芦那韦/利托那韦合用时,推荐阿托伐他汀的开始剂量为10mg q.d.,根据临床反应逐渐增加阿托伐他汀的剂量。
达芦那韦/利托那韦(600/100mg b.i.d.)可使单次剂量普伐他汀(40mg)的暴露量增加大约80%,但又在部分受试者中发生。当需要将普伐他汀与本品/利托那韦联用时,推荐普伐他汀以可能的最小剂量开始,并逐渐滴定至满意的临床效果,同时,应监测其安全性。
H2-受体拮抗剂和质子泵抑制剂
奥美拉唑(20mg q.d.)或雷尼替丁(150mg b.i.d.)与达芦那韦/利托那韦(400/100mg b.i.d.)合用时,不影响达芦那韦的暴露量。基于这些结果,达芦那韦/利托那韦可与H2受体拮抗剂及质子泵抑制剂同服,而无需调整剂量。
免疫抑制剂(环孢霉素、他克莫司、西罗莫司)
与达芦那韦/利托那韦同服时,环孢霉素、他克莫司、西罗莫司的暴露量增加。与达芦那韦/利托那韦同服时,推荐对免疫抑制剂进行监测。
酮康唑、伊曲康唑和伏立康唑
酮康唑、伊曲康唑和伏立康唑都是CYP3A4的强力抑制剂和底物。全身使用酮康唑、伊曲康唑或伏立康唑和达芦那韦/利托那韦时,可增加达芦那韦的血浆浓度;同时,达芦那韦/利托那韦也可增加酮康唑、伊曲康唑或伏立康唑的血浆浓度。在一项相互作用试验中证实,酮康唑(200mg b.i.d.)和达芦那韦/利托那韦(400/100mg b.i.d.)同时使用时,酮康唑和达芦那韦的暴露量分别增加212%和42%。当需要同服时,酮康唑或伊曲康唑每天的剂量不应超过200mg。尽管伏立康唑的代谢也涉及CYP3A4之外的酶,当与达芦那韦/利托那韦同服时,伏立康唑的暴露量预期会增加。
美沙酮
一项研究本品/利托那韦(600/100mg b.i.d.)对稳定美沙酮维持治疗影响的相互作用试验表明,R-美沙酮AUC降低16%。根据药代动力学和临床试验结果,当美沙酮与本品/利托那韦同服时,无需进行剂量调整。然而,因为可能需要对一些患者进行维持治疗调整,因此应进行临床监测。
雌激素为基础的避孕药物
本品/利托那韦(600/100mg b.i.d.)与炔雌醇和炔诺酮的相互作用试验结果表明,稳态时对炔雌醇和炔诺酮的暴露分别降低44%和14%。可使用其他非激素类避孕药进行替代。
PDE-5抑制剂
在一项相互作用试验中,以西地那非100mg单剂作为对照,比较了西地那非25mg单剂与达芦那韦/利托那韦(400/100mg b.i.d.)合用时西地那非的全身暴露量。应谨慎同时使用PDE-5抑制剂和达芦那韦/利托那韦。如果有指征需要同时使用达芦那韦/利托那韦与西地那非、伐地那非或他达拉非时,推荐西地那非48小时内单次剂量不超过25mg,伐地那非72小时内单剂量不超过2.5mg或他达拉非72小时内单次剂量不超过10mg。
利福布丁(利福布汀)
利福布丁(利福布汀)是CYP450酶的底物。在一项有关药物相互作用的试验中,达芦那韦/利托那韦(600/100mg b.i.d.)与利福布汀(150mg隔日一次[q.o.d.])联用时,达芦那韦的全身暴露增加了57%。基于本品/利托那韦的安全性特点,达芦那韦暴露在利福布汀存在时所出现的增加并不需要对本品/利托那韦进行剂量调整。相互作用试验表明,利福布汀在以300mg q.d.单用和以150mg q.o.d.与本品/利托那韦(600/100mg b.i.d.)联用时的全身暴露是相当的,其活性代谢产物25-O去乙酰利福布汀的暴露增加。在接受二者联用的患者中,应将利福布汀的常规剂量300mg/day降低75%(即利福布汀150mg q.o.d.),同时加强对利福布汀相关不良事件的监测。
选择性5-羟色胺再摄取抑制剂(SSRIs)
帕罗西汀(20mg q.d.)或舍曲林(50mg q.d.)与达芦那韦/利托那韦(400/100mg b.i.d.)之间的相互作用试验中,帕罗西汀或舍曲林不影响达芦那韦的暴露量。达芦那韦/利托那韦存在时,舍曲林和帕罗西汀的暴露量分别降低49%和39%。如果SSRIs和达芦那韦/利托那韦同服时,应根据抗抑郁药物应答的临床评估,对SSRIs的剂量进行仔细摸索。另外,帕罗西汀或舍曲林剂量稳定的患者在使用达芦那韦/利托那韦初期,应监测抗抑郁药物的应答。
药物过量
达芦那韦/利托那韦急性过量用药经验有限。达芦那韦口服液3200mg或片剂1600mg单次剂量与利托那韦联合使用时,在健康志愿者中没有发现明显不良反应。
达芦那韦过量时没有特殊的解毒药。达芦那韦过量的治疗主要是一般支持疗法,包括对患者的生命体征监测和临床状态的观察。如果有指征,可通过呕吐或胃部灌洗来清除未吸收的活性药物。也可使用活性炭去除未吸收的活性药物。由于达芦那韦与蛋白质高度结合,透析对活性药物的完全清除可能没有帮助。
临床试验
本品/利托那韦疗效的证据来自对两项正在进行的随机、对照试验的24周结果分析,这两项试验的受试者均为曾经有接受抗病毒治疗经历的HIV-1成人患者(POWER1和POWER2试验)。这些结果又被两项开放标签试验TMC114-C215和TMC114-C208的24周汇总分析所证实(POWER3分析)。
POWER1和POWER2是针对高水平PI耐药患者的随机、对照、IIb期临床试验,每项试验由两部分组成:第一部分为部分设盲、剂量摸索;第二部分的期限较长,所有随机分配到本品/利托那韦治疗组的患者,推荐剂量为600/100mg b.i.d.。
这些试验中,受试者的入选条件包括,血浆HIV-1RNA>1000copies/ml,曾经接受蛋白酶抑制剂、非核苷类及核苷类逆转录酶抑制剂治疗,在筛查期至少携带一种PI主要突变,在筛查时稳定服用含有PI的方案至少8周。根据PI突变的数目、筛查时的病毒载量和是否使用恩夫韦肽进行随机分组。分析的对象包括318例POWER1试验中和319例POWER2试验中完成24周治疗或提前中止的患者。
本品/利托那韦与对照组之间人口统计学和基线特征无显著性差异。在两项试验中,共有131例患者服用本品/利托那韦600/100mg b.i.d.,平均年龄为43.0岁(范围27~73岁),其中男性89%,百人81%,黑人10%,希伯来人7%;平均基线血浆HIV-1RNA为4.61log10 copies/ml(范围是2.99~6.44log10 copies/ml);平均基线CD4细胞计数为153×106/l(范围是3~776×106/l);达芦那韦FC的平均值为4.3。在本品/利托那韦600/100mg b.i.d.治疗组,患者曾经服用的抗病毒药物中平均4种蛋白酶抑制剂、5种核苷类逆转录酶抑制剂和1种非核苷类逆转录酶抑制剂,而对照组为4种蛋白酶抑制剂、6种核苷类逆转录酶抑制剂和1种非核苷类逆转录酶抑制剂;本品/利托那韦组中20%的患者曾经使用恩夫韦肽,而对照组仅为17%。
与基线相比病毒载量下降1.0log10定义为病毒学应答。治疗组接受的药物为本品/利托那韦+优化基础药物组合(OBR),而对照组接受的药物为研究者选定的蛋白酶抑制剂+OBR,OBR由至少两种核苷类药物伴或不伴恩夫韦肽组成。在第24周时评价了两组的病毒学疗效。基于耐药试验和既往的用药史,对照组选用的蛋白酶抑制剂包括:洛匹那韦/利托那韦(36%),安普那韦或夫沙那韦(34%),沙奎那韦(35%)和阿扎那韦(17%)。
对照组中23%的患者使用增效的双蛋白酶抑制剂,所有患者中47%的患者使用恩夫韦肽,其中35%的患者是首次应用恩夫韦肽。
POWER3:其他的本品/利托那韦600/100mg b.i.d.的疗效数据是来自针对曾经接受治疗的患者的两项非随机试验TMC114-C215和TMC114-C208。试验共有246例患者纳入24周POWER3疗效分析,治疗采用本品/利托那韦,剂量为600/100mg b.i.d.,OBR由至少两种核苷类药物伴或不伴恩夫韦肽组成。入选标准与POWER1和POWER2相同,基线特征与POWER1和POWER2具有可比性。基线时血浆HIV-1RNA的中位值为4.60log10 copies/ml(范围是1.69~6.43log10 copies/ml),CD4细胞计数的中位值是115×106/l(范围是0~831×106/l),达芦那韦FC的中位值是3.2。患者曾经暴露于5种PIs、6种NRTIs和1种NNRTI,30%的患者曾经使用恩夫韦肽。基线特征基于TMC114-C215和TMC114-C208收录的327例患者,而疗效分析则基于完成24周治疗或提前退出的246例患者的中期数据。
下表显示的是POWER1和POWER2试验以及POWER3对采用品/利托那韦600/100mg b.i.d.治疗24周的疗效结果
http://x1.webres.medlive.cn/drugref/ChemPreparationDetail/201511231128480592.jpg
1)未完成治疗者退定为治疗失败:提前退出者病毒载量的变化推定为0.
2)统计处理结果基于最小二乘方,来源于含有分层因素的ANOVA模型,P值<0.001.
3)末次观察前推法
4)根据TLOVR分析法进行推算
5)奇数比来源于包含分层因素的逻辑回归模型,各分组间应答率和变化基于已观察数据,P值<0.001.
在POWER1和POWER2汇总分析中,本品/利托那韦(600/100mg b.i.d.)治疗组病毒载量对数值较基线时下降患者的比例明显超过对照组。在第24周时,病毒载量至少降低1.0log10的患者在本品/利托那韦治疗组所占的比例为70%,而对照组为21%;本品/利托那韦组中HIV-1RNA<50copies/ml的患者占45%,对照组为12%。
POWER3的24周疗效分析证实了在POWER1和POWER2试验中所见的病毒载量下降和CD4+细胞计数增加。在纳入24周分析的246例患者中,65%出现病毒学应答(血浆病毒载量较基线时至少下降1.0log10),40%病毒载量达到50copies/ml以下。
此外,本品/利托那韦600/100mg b.i.d.长期疗效的数据来源于POWER1 and POWER2 试验对经治患者48周治疗的汇总分析。对于治疗48周或提前退出的患者,48周汇总分析的结果显示,病毒学反应依然良好,在24周和48周时同样比例的患者病毒载量达到检测不到的水平(HIV RNA<50copies/ml)(分别为45%和45%)。
本品/利托那韦对经治患者的抗病毒活性
POWER1和POWER2 II期试验和POWER3分析中,458例经历广泛抗病毒治疗的患者接受本品/利托那韦治疗,采用的剂量为600/100mg b.i.d.。
本品/利托那韦治疗过程中对体内耐药病毒的选择
在POWER1和POWER2 II期试验和POWER3分析中,受试者为高度经治的患者,接受本品/利托那韦治疗。无论是病毒载量反弹(50例),还是病毒载量从未得到有效抑制(70例),均为病毒学失败的患者,从这些患者中分离的蛋白酶多重耐药HIV-1病毒株的分析显示,这些病毒株出现与达芦那韦敏感性降低有关的氨基酸替换。在经本品/利托那韦治疗病毒学失败的患者标本中分离的病毒株中,氨基酸替换大于20%的位点为V32I和I54L,在10~20%之间者为L33F、I47V和L89V。
与其他蛋白酶之间的体内交叉耐药
目前关于本品/利托那韦选择的病毒株的交叉耐药情况所知甚少。对于本品/利托那韦100/600mg b.i.d.治疗组病毒反跳患者中分离的病毒株,与基线相比,终点时本品FC却几乎没有增加(平均增加0.82),提示这两种蛋白酶抑制剂之间交叉耐药的程度有限。基线时对替拉那韦耐药的患者(FC>3),在24周时,病毒载量的变化的平均值为-1.38log10。由于在基线时病毒已经对其他蛋白酶抑制剂出现耐药,因而无法研究这些药物的FC增加。对于基线时无敏感蛋白酶抑制剂可用的患者(不包括替拉那韦),24周时病毒载量变化的平均值为-1.57log10。
基线时基因型耐药或表型耐药及病毒学检测结果
通过分析数据,评价了基线存在的一些特定蛋白酶抑制剂(PI)耐药突变对病毒学疗效的影响。在POWER1和POWER2试验中,基线时存在的V32I、I47V或I54L/M突变与达芦那韦的病毒学疗效下降和敏感性降低有关。此外,基线时存在7个以上PI耐药相关突变(在30、32、36、46、47、48、50、53、73、82、84、88或90位点出现任何改变)的患者病毒学疗效极差。然而,与对照组相比,达芦那韦/利托那韦治疗的各个亚组(基线时根据突变的类型和数量分组)发生病毒学应答的比例通常较高。
在POWER1和POWER2辅助分析和POWER3分析中,基线时存在以下3种或以上突变与PREZISTA/rtv病毒学疗效降低有关,这些突变包括V11I、V32I、L33F、I47V、I50V、I54L/M、G73S、L76V、I84V和L89V。
根据基线时基因型*,本品/rtv600/100mg b.i.d.的病毒学疗效:POWER1、POWER2和POWER3处理分析的结果
http://x1.webres.medlive.cn/drugref/ChemPreparationDetail/201511231131490163.jpg
*与本品/rtv病毒学疗效极差有关的突变的数量(V11I、V32I、L33F、I47V、I50V、I54L/M、G73S、L76V、I84V和L89V)
特定突变或突变类型与疗效相关性的结论可能会随着进一步研究的数据而发生变化。
基线时达芦那韦的表现型(相对于参照而发生的敏感性变化)是病毒学疗效的预测因素。
根据基线时达芦那韦表现型而评价的病毒学应答比例见下表。这些基线时表型分组基于POWER1、POWER2试验和POWER3分析入选的病人人群,并不代表全面的临床敏感性数据。基于PI经治患者治疗前对达芦那韦的敏感性,这些数据向临床医师提供了病毒学成功可能性的信息。
根据基线时表型,本品/rtv 600/100mg b.i.d.的病毒学疗效:POWER1、POWER2和POWER3处理分析的结果
对于曾经抗病毒治疗失败的患者,在选择新的治疗方案时,在可能的情况下,应当慎重考虑治疗史以及耐药检测的结果。
药理毒理
药理作用
作用机理
达芦那韦是一种HIV-1蛋白酶抑制剂,选择性抑制病毒感染细胞中HIV编码的Gag-Pol多蛋白的裂解,从而阻止成熟的感染性病毒颗粒的形成。
达芦那韦与HIV-1蛋白酶紧密结合,Kp值为4.5×10-12M。达芦那韦对于蛋白酶抑制剂耐药相关的突变(RAM)具有一定的疗效,但达芦那韦对目前检测到的13种人体细胞蛋白酶没有抑制作用。
体外抗逆转录病毒活性
在急性感染的T细胞系,人外周血单核细胞和人单核/巨噬细胞中,达芦那韦具有抗HIV-1实验室株和临床分离株以及HIV-2实验室株和临床分离株的活性,EC50的范围在1.2~8.5nM(0.7~5.0ng/ml)。达芦那韦在体外具有广谱抗HIV-1活性,对于M组(A、B、C、D、E、F、G亚型)和O组原始分离株均具有抗逆转录病毒活性,EC50的范围在<0.1~4.3nM,这些值显著小于50%细胞毒浓度范围(87μM~>100μM)。
在人血清中,达芦那韦的EC50增加的中位因子为5.4。达芦那韦与蛋白酶抑制剂利托那韦、奈非那韦、或安普那韦联合使用时具有协同作用,与蛋白酶抑制剂茚地那韦、沙奎那韦、洛匹那韦、阿扎那韦、或替拉那韦,核苷(酸)类逆转录抑制剂齐多夫定、拉米夫定、扎西他滨、去羟肌苷、司他夫定、阿巴卡韦、恩曲他滨或替诺福韦,非核苷类逆转录酶抑制剂etravirine、奈韦拉平、地拉韦啶或依非韦伦,以及融合抑制剂恩夫韦肽联合使用时具有相加作用。没有发现达芦那韦与其他抗逆转录病毒药物之间具有拮抗作用。
体外耐药
HIV-1体外试验中,从野生株中选择出达芦那韦耐药株是一个漫长的过程(长于2年),在达芦那韦浓度大于200nM时,选择的病毒株不能生长。这些选择性病毒株对达芦那韦的敏感性降低(范围6~21倍),蛋白酶序列发生3~6个氨基酸替换,目前正在鉴定和研究对达芦那韦敏感性降低的耐药决定簇。
对于携有多PI耐药相关突变的9个HIV病毒株,体外药物选择试验显示,达芦那韦耐药HIV-1(EC50的变化范围:53~641倍)在蛋白酶序列出现22处突变,9例达芦那韦耐药分离株中L10F、V32I、L33F、S37N、M46I、I47V、I50V、L63P、A71V和I84V出现的频率在50%以上。对达芦那韦耐药(倍数变化[FC]>10)的HIV-1株,其蛋白酶序列上至少需要8个达芦那韦体外选择的突变,其中在选择前蛋白酶序列上已存在至少2个突变。
在对安普那韦、阿扎那韦、茚地那韦、洛匹那韦、奈非那韦、利托那韦、沙奎那韦和/或替拉那韦耐药的1113例临床分离株,以及POWER1、POWER2和POWER3试验收录的886例基线分离株中,只有携带10个以上PI耐药相关突变的病毒株对达芦那韦耐药的平均倍数变化(FC)才大于10。
体外交叉耐药
蛋白酶抑制剂之间存在交叉耐药。对安普那韦、阿扎那韦、茚地那韦、洛匹那韦、奈非那韦、利托那韦、沙奎那韦和/或替拉那韦耐药的3309例临床分离株中,90%对达芦那韦的敏感性下降<10倍,这说明对大多数PI耐药的病毒株对达芦那韦仍然敏感。
从PI耐药病毒株中选择的9例对达芦那韦耐药的病毒株中7例具有替拉那韦的表型耐药试验资料,其中6例替拉那韦EC50的倍比变化小于3,提示这两种蛋白酶抑制剂之间交叉耐药的程度有限。
达芦那韦与核苷(酸)类逆转录酶抑制剂、非核苷酸类逆转录酶抑制剂、侵入抑制剂或整合酶抑制剂之间不可能存在交叉耐药,因为达芦那韦与这些抑制剂之间作用于病毒的靶位不同。
毒理研究
达芦那韦单药的动物毒性试验已在小鼠、大鼠和犬中进行,与利托那韦合用的毒性试验已在大鼠和犬种进行。
在大鼠和犬慢性毒性试验中,达芦那韦仅有局限的毒副作用。在大鼠试验中,暴露量在100mg/kg/天或以上和低于临床暴露水平时,药物作用的主要靶器官为造血系统、凝血系统、肝脏和甲状腺。许多红细胞相关的参数出现小幅下降,伴有激活的PTT(部分促凝血酶原激酶时间)延长。肝脏和甲状腺的变化可能是对大鼠酶诱导作用的适应性反应,而非不良反应。在与利托那韦合用药的毒性试验中,在大鼠中没有关于其它靶器官毒性的报道。高达120mg/kg/天的剂量以及在临床推荐剂量的暴露量之下,在犬种没有发现严重的毒性反应或靶器官损害。
大鼠试验显示,达芦那韦剂量达1000mg/kg/天时对交配和繁殖没有影响,暴露水平(AUC-0.5 fold)低于人体临床推荐的治疗量。在同样剂量水平下,达芦那韦单药对大鼠和兔没有致畸作用,与利托那韦合用时对小鼠也没有致畸作用,暴露水平低于人体临床推荐的剂量。在大鼠出生前后的发育评估试验中,达芦那韦无论是否与利托那韦联合均可引起哺乳期子鼠体重的短期下降,这可能与通过乳汁导致的药物暴露有关,在断奶后,药物对子鼠的各项功能没有影响。
在直接接受达芦那韦(从20mg/kg至1000mg/kg)的年龄在23天至25天的幼年大鼠中观察到了死亡的现象,同时,在一些动物中,还观察到惊厥的现象。在这个年龄范围内,血浆、肝脏和大脑中的暴露量取决于剂量和年龄,同时,该暴露量大大高于在成年大鼠中观察到的暴露量。这些结果的出现与达芦那韦代谢涉及的CYP450肝酶的个体发生与脑血屏障不成熟有关。年龄为26天的幼年大鼠接受1000mg/kg达芦那韦(单次给药)或年龄为23至50天的幼年大鼠接受500mg/kg(重复给药)时,未观察到治疗相关死亡,同时,其暴露和毒性特点与成年大鼠中所观察到的特点相当。在人体中,药物代谢酶的活性在3岁时即已达成年时的数值。
通过管饲法给药(最多104周)评价了达芦那韦在小鼠和大鼠中的致癌性。小鼠接受的每日剂量包括150、450和1000mg/kg,而大鼠接受的每日剂量包括50、150和500mg/kg。在两个物种的雌性和雄性中均观察到肝细胞腺瘤和肝细胞癌的发生率随剂量的增加而增加。在雄性大鼠中观察到了甲状腺滤泡细胞腺瘤。达芦那韦给予小鼠或大鼠后并未导致其他任何良性或恶性肿瘤的发生率出现具有显著统计学意义的增加。所观察到的在啮齿类动物中的肝细胞结果被认为与人类的相关性有限。重复给予大鼠达芦那韦可诱导肝微粒体酶并增加甲状腺激素的清除,这些会导致大鼠(但非人类)更易发生甲状腺肿瘤。在所测试的最高剂量下,达芦那韦的系统暴露(基于AUC)分别是人接受推荐治疗剂量(600/100mg、每天两次或800/100mg、每天一次)时所观察到的暴露的0.4至0.7倍(小鼠)和0.7至1倍(大鼠)。达芦那韦在细菌回复突变试验(Ames)、在人淋巴细胞内的染色体畸变以及在小鼠体内的微核试验中,结果均为阴性。
药代动力学
本品与利托那韦合用的药代动力学特点,在健康成人志愿者和HIV-1感染者中已获得评估。HIV-1感染者达芦那韦的暴露量高于健康受试者,其原因可能在于HIV-1感染者α-1-酸性糖蛋白(AAG)的浓度较高,达芦那韦与血浆AAG结合较多,从而导致达芦那韦血浆浓度较高。
达芦那韦主要通过CYP3A4代谢,利托那韦可抑制CYP3A4的活性,因此,在与利托那韦合用时,达芦那韦的血浆浓度会显著增加。
吸收
口服后,达芦那韦快速吸收。与低剂量利托那韦同服时,达芦那韦的最大血浆浓度通常在服药后2.5~4.0小时达到。
600mg单剂量本品的绝对口服生物利用度大约为37%,与利托那韦100mg b.i.d.合用时,生物利用度增加到82%。在利托那韦的作用下,达芦那韦的全身暴露量增加约14倍。
与进餐时服用相比,不与食物同服时本品与低剂量利托那韦合用的相对生物利用度降低30%,因此,本品片剂应当与利托那韦和食物同服,食物的类型不影响达芦那韦的暴露量。
分布
约95%的达芦那韦与血浆蛋白结合,主要与血浆α-1-酸性糖蛋白结合。
代谢
人肝微粒体(HLM)的体外试验显示,达芦那韦主要进行氧化代谢,被肝脏CYP系统广泛代谢,绝大多数被CYP3A4同工酶代谢。一项在健康志愿者中进行的14C-达芦那韦试验显示,单次服用本品/利托那韦400/100mg后血浆中大部分放射活性物质来源于达芦那韦本身,在人体内已发现至少3种氧化代谢产物,这些产物对于野生型HIV的活性小于达芦那韦活性的至少10倍。
清除
单次服用400/100mg 14C-达芦那韦/利托那韦后,粪便和尿液中检测的14C-达芦那韦大约分别占79.5%和13.9%。达芦那韦原药大约分别占粪便和尿液中剂量的41.2%和7.7%。与利托那韦合用时,达芦那韦的终末清除半衰期约为15小时。达芦那韦(150mg)单独用药以及与低剂量利托那韦合用是,静脉清除率分别为32.8l/h和5.9l/h。
特殊人群
儿童
本品与利托那韦合用在儿童受试者中的药代动力学尚在研究阶段,目前没有关于儿童的推荐剂量。
老年人
人群药代动力学分析显示,HIV感染人群(n=12,年龄≥65岁)本品的药代动力学在18~75岁这一年龄范围内并无明显差异(见注意事项部分)。
性别
人群药代动力学分析显示,女性达芦那韦的暴露量略高于男性(16.8%),这一差异无临床意义。
肾功能障碍
14C-达芦那韦/利托那韦的质量守恒试验的结果显示,大约7.7%的达芦那韦以原型从尿中排出。
尽管本品没有在肾功能障碍的患者中进行研究,人群药代动力学分析显示,本品的药代动力学在中度肾功能障碍(肌酐清除率在30~60ml/分钟,n=20)的HIV患者中没有显著影响(见用法用量及注意事项部分)。
肝功能障碍
在一项针对本品/利托那韦(600/100mg)、每日两次的多次给药研究结果表明,达芦那韦在轻度(Child Pugh A级,n=8)和中度(Child Pugh B级,n=8)肝损伤受试者体内的稳态药代动力学参数与其在健康受试者的参数相当。尚未研究严重肝功能损伤对达芦那韦的药代动力学影响(见用法用量及注意事项部分)。
贮藏
低于30℃保存。
包装
高密度聚乙烯塑料瓶装,配有儿童不易打开的瓶盖。120片/瓶/盒。
有效期
24个月
执行标准
JX20070057