

赛沃替尼详细说明书
【药品名称】
通用名称:赛沃替尼片
商品名称:沃瑞沙/ORPATHYS\"
英文名称: Savolitinib Tablets
汉语拼音: Saiwotini Pian
【成份】
本品主要成分为赛沃替尼。
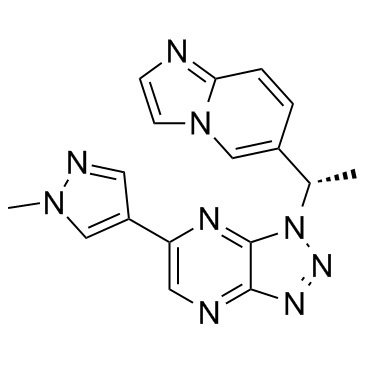
化学名称: 1-(1S)-1-(咪唑并[1,2-a]吡啶 6-基)乙基]-6-(1-甲基1H-吡唑: 4基)-1H-[1,2,3]三唑并[4,5-b]吡嗪
分子式: CiHisN,
分子量: 345.36
【性状】
薄膜衣片,除去包衣后显白色至黄色。
【适应症】
【用法用量】
推荐剂量和服用方法
对于体重>=50公斤的患者,建议起始剂量为600 mg,每日一次口服,直到疾病进展或出现不可耐受的毒性。
对于体重<50公斤的患者,建议起始剂量为400 mg,每日一次口服,直到疾病进展或岀现不可耐受的毒性。
建议每日相同时段在餐后即刻服用本品。
剂量调整
医生应在患者用药过程中密切监测,根据患者个体的安全性和耐受性调整用药,包括暂停本品、降低剂量或永久停用本品。
本品的剂量调整建议参见表1,不良反应导致治疗调整的建议参见表2。
| 剂量水平 | 赛沃替尼每日口服剂量 | |
| 起始剂量 | 600 mg每日一次 (体重>=50 kg) |
400 mg每日一次 (体重<50 kg) |
| 剂量水平-1 (第一次减量) | 400 mg每日一次 | 300 mg每日一次 |
| 剂量水平-2 (第二次减量) | 300 mg每日一次 | 200 mg每日一次 |
| 剂量水平-3 (第三次减量) | 200 mg每日一次 | - |
| 不良反应 | 赛沃替尼治疗调整 |
| 肝毒性 | |
| • ALT或AST>8xULN且合并TBIL升高超过基线水平或ULN • ALT或AST>5xULN持续>2周且合并TBIL 升高超过基线水平或ULN • ALT 或 AST>3xULN 并且(TBIL>2xULN 或INR>1.5,在未使用升高INR的抗凝剂的情况下) • AST或ALT>3xULN伴明显乏力、恶心、 呕吐、上腹痛或腹胀、发热、皮疹和/或嗜酸性粒细胞>5% |
• 永久停用本品 |
| 不良反应 | 赛沃替尼治疗调整 |
| • 再次发生ALT或AST>5xULN • 再次发生 ALT或 AST>3〜5xULN且合并 TBIL>1.5~2xULN |
|
| • ALT或AST>5xl)LN且TBIL不高于基线水平或ULN • ALT 或 AST>3xULN, TBIL 升高至 1.5〜 2xULN • 再次发生ALT或AST>3〜5xULN但 TBIL不高于基线水平或ULN |
・ 暂停本品,重复肝功能检测,每周两次 > 如果在1周内恢复至1级(接近基线水平)或基线水平,下调1个剂量水平用药,重复肝功能检测6周,每周两次 >否则,永久停用本品 |
| 过敏反应 | |
| ・急性严重过敏反应或严重速发过敏反应 (包括速发过敏反应性休克) | • 永久停用本品 |
| ・其他过敏反应 | ・建议医生的指导下尽快开始抗过敏治疗,同时: >永久停用 或 >暂停本品,直至病情恢复。只有在医生认为继续使用本品的受益超过风险、并预防使用抗过敏治疗至少24小时的前提下,才可下调1个剂量水平恢复用药(请参考 【注意事项】) |
| 其他 | |
| ・ CTCAE 3级或4级毒性 | ・ 暂停本品,直至恢复至1级,下调1个剂量水 平恢复用药 ・否则,永久停用本品 |
【特殊患者人群】
肝功能不全轻度肝功能不全的患者(总胆红素<ULN和ALT或AST>ULN;或1.5XULN>总胆红素>ULN以及任意水平ALT或AST)服用本品无需调整起始剂量。目前尚无中度和重度肝功能不全患者(总胆红素>1.5xULN以及任意水平ALT或AST)的研究数据,因此中重度肝功能不全患者应在医生指导下慎用本品,并严密监测其肝功能。
肾功能不全
轻度和中度肾功能不全患者服用本品无需调整起始剂量。目前尚无重度肾功能不全患者的研究数据,重度肾功能不全患者应在医生指导下谨慎服用本品,并严密监测其肾功能。
儿童患者
尚无本品用于18岁以下儿童或青少年患者的临床数据。
老年患者
年龄>=65岁患者无需调整起始剂量。
【不良反应】
本说明书描述了在临床研究中观察到的判断为可能由本品引起的不良反应及其近似的发生率。由于临床研究是在各种不同条件下进行的,在一个临床研究中观察到的不良反应的发生率不能与另一个临床研究观察到的不良反应发生率直接比较,也可能不能反映临床实践中的实际发生率。赛沃替尼的安全性数据来自于5项临床试验,总计有345例肿瘤患者接受本品单药治疗,其中有338例患者[含关键性II期研究的70例MET外显子14跳变的非小细胞肺癌患者(肺肉瘤样癌和其他非小细胞肺癌)]暴露于推荐剂量及以上(>=400 mg,每日一次) 剂量水平。在接受>=400 mg剂量的患者中,20.4%的患者因不良反应而暂停治疗;导致暂停治疗的不良反应(>=1%)为水肿(4.7%)、恶心(3.6%)、呕吐(3.6%)、发热 (3.8%)、天门冬氨酸氨基转移酶升高(2.4%)、丙氨酸氨基转移酶升高(2.1%)、疲乏/乏力(1.8%)、食欲减退(1.5%)、贫血(1.5%)、皮疹(1.5%),以及肝功能异常
(1.2% )。 15.4%的患者因不良反应而减量;导致减量的不良反应(>=1%)为水肿 (4.4%)、丙氨酸氨基转移酶升高(3.6%)、天门冬氨酸氨基转移酶升高(3.3%)、恶 心(1.5%)、疲乏/乏力(1.2%)、发热(1.2%),以及肝功能异常(1.2%) 。 11.8%的患者(40例)因不良反应而永久停药;导致永久停药的不良反应(>=1%)为肝功能异常
(3.8%)、呕吐(1.5%)、丙氨酸氨基转移酶升高(2.1%),疲乏/乏力(1.2%)、水肿 (1.2%)、天门冬氨酸氨基转移酶升高(1.2%),以及严重过敏反应(1.2%)。
在接受>=400 mg剂量的患者中,常见(>=10%)不良反应为恶心(44.7%)、水肿 (40.5%)、疲乏/乏力(31.1%)、呕吐(31.1%)、食欲减退(21.0%)、低白蛋白血症(17.2%)、贫血(16.6%)、发热(15.7%)、腹泻(13.6%),以及肝功能异常(11.8%) 。
常见(>=10%)实验室检查异常包括天门冬氨酸氨基转移酶升高(18.0%)和丙氨酸氨基转移酶升高(16.3%)。
在接受>=400 mg剂量的患者中发生的不良反应详见表3。
| 不良反应 | 赛沃替尼(>=400 mg)(N=338) | |
| 所有级别(%) | >=3 级(%) | |
| 胃肠系统疾病 | ||
| 恶心 | 44.7 | 1.5 |
| 呕吐1 | 31.1 | 1.5 |
| 腹泻2 | 13.6 | 0.6 |
| 全身性疾病及给药部位各种反应 | ||
| 不良反应 | 赛沃替尼(>=400 mg) (N=338) |
|
| 所有级别(%) | >=3 级(%) | |
| 水肿3 | 40.5 | 3.8 |
| 疲乏/乏力 | 31.1 | 4.4 |
| 发热4 | 15.7 | 1.2 |
| 代谢及营养类疾病 | ||
| 食欲减退 | 21.0 | 1.2 |
| 低白蛋白血症5 | 17.2 | 1.2 |
| 血液及淋巴系统疾病 | ||
| 贫血6 | 16.6 | 3.3 |
| 肝胆系统疾病 | ||
| 肝功能异常7 | 11.8 | 5.6 |
| 皮肤及皮下组织类疾病 | ||
| 皮疹8 | 9.5 | 0.6 |
| 免疫系统疾病 | ||
| 严重超敏反应9 | 1.5 | 1.2 |
| 各类检查 | ||
| 天门冬氨酸氨基转移酶升高 | 18.0 | 6.2 |
| 丙氣酸氨基转移酶升高 | 16.3 | 5.9 |
| 血碱性磷酸酶升高 | 7.1 | 0 |
| 血胆红素升高 | 7.1 | 0.6 |
| γ-谷氨酰转移酶升高 | 4.7 | 1.2 |
| 心电图QT间期延长 | 3.0 | 0 |
1. 呕吐包括呕吐、干呕;
2. 腹泻包括腹泻、排便频率增加;
3. 水肿包括外周水肿、水肿、面部水肿、外周肿胀、肿胀、局部水肿、眼睑水肿、全身性水肿、阴囊水肿、睾丸水肿、牙龈肿胀、眼水肿、生殖器水肿、阴茎水肿;
4. 发热包括发热、流感样症状、寒战、体温升高、髙热;
5. 低白蛋白血症包括血白蛋白降低、低白蛋白血症;
6. 贫血包括贫血、血红蛋白降低、红细胞计数下降;
7. 肝功能异常包括肝功能异常、药物诱导的肝损伤、肝损伤、肝病损、肝性脑病、肝脏毒性、肝脏 疾病;
8. 皮疹包括皮疹、斑丘疹、药疹、丘疹、红斑性发疹、瘙痒性皮疹、水泡疹、瘗疮样皮炎、过敏性 皮炎、皮炎、多形性红斑、中毒性皮疹;
9. 严重超敏反应包括药物性超敏反应、速发过敏反应性休克、超敏反应。
【特定的不良反应】
肝毒性在接受>=400 mg剂量的患者中,肝毒性主要表现为天门冬氨酸氨基转移酶升高、丙氨酸氨基转移酶升高、低白蛋白血症、肝功能异常、血胆红素升高、碱性磷酸酶升高等肝功能检查异常,大多数为1〜2级;发生率>1.5%的>=3级的肝毒性事件包括天门冬氨酸氨基转移酶升高(6.2%)、丙氨酸氨基转移酶升高(5.9%)、肝功能异常(5.6%)。从开始服药到不良反应发生的中位时间为31天。另外,在肝毒性事件中,药物诱导的肝损伤(DILI)(所有级别)的发生率为1.2%, >=3级的DILI的发生率为0.9%。在所有肝毒性事件中,有1例(0.3%)致死性病例。
发热
在接受>=400 mg剂量的患者中,发热相关事件主要表现为发热、寒战、高热和流感样症状等,从开始服药到不良反应发生的中位时间为20天,大部分为1〜2级,没有死亡的病例报告。
严重过敏反应
在接受>=400 mg剂量的患者中,符合严重不良事件标准的过敏反应包括药物性超敏反应(0.9%)、速发过敏反应性休克(0.3%)、超敏反应(0.3%),从开始服药到不良反应发生的中位时间为15天,没有死亡病例报告。
水肿
在接受>=400 mg剂量的患者中,水肿相关事件包括外周水肿、水肿、面部水肿、外周 肿胀、肿胀等,从开始服药到不良反应发生的中位时间为50天,大多数为1〜2级;发生率>1.5%的3级水肿事件仅包括外周水肿(2.4%),没有死亡病例报告。
【禁忌】
有本品严重过敏史者或对本品任何成分过敏者禁用。妊娠、哺乳期妇女禁用。
【注意事项】
肝毒性临床研究中观察到本品可能引起肝功能检查异常和药物诱导的肝损伤等,多为1〜2 级,有1例(0.3%)致死性病例。详情可参见“特定不良反应”。
临床研究中从开始接受本品至发生肝毒性的中位时间为31天(范围1〜420天),经过保肝治疗以及剂量调整或暂停用药后,通常可恢复至=<1级或用药前水平。
使用本品前存在肝脏功能异常风险因素(如肝胆疾病、肝转移、肝功能异常等)的患者需要全面谨慎地评估。治疗期间应定期监测肝功能(如转氨酶及血胆红素),建议开始用药后前3个月内每周监测。3个月后可根据肝功能检查的结果调整监测频率,如每 2-3周评估肝功能,必要时调整剂量或停用本品。(参见【用法用量】中的“剂量调整”)
严重过敏反应
临床研究中,从开始接受本品至发生严重过敏反应的中位时间为15天(范围10〜90 天)。
临床研究中观察到本品可引起发热、寒战和流感样症状,从开始接受本品至发热的 中位时间为20天(范围1〜620天)。
本品引起超敏反应(表现为一系列症状,包括但不限于:药物相关性发热、皮肤过敏反应、肝酶升高、血细胞下降、肌痛/关节痛)。这些反应可在用药后数天至数周内发生,但大多发生在用药后六周内。
临床研究中观察到某些开始表现为超敏反应的患者短期停药后,当恢复本品治疗时会出现急性严重超敏反应,包括速发过敏反应。
患者疑似发生本品相关超敏反应(排除已确认的感染病因)时,应根据患者具体病情给予相应的治疗(如抗组胺药、糖皮质激素、退热药等)并停用本品。待症状恢复后,只有在医生认为继续使用本品的受益超过风险时方可减量用药。在恢复本品前至少24小时开始预防和伴随应用抗过敏药物(如糖皮质激素和抗组胺药),恢复本品用药当天必须在院内留观24小时,恢复本品用药后需继续使用抗过敏药物至少1周,并由医生判断是否仍需继续使用抗过敏药物。一旦发生急性超敏反应或速发过敏反应,必须立即进行医学干预,并应永久停用本品,参见【用法用量】。
【孕妇及哺乳期妇女用药】
必须告知育龄女性本品可能伤害胎儿。育龄女性服用本品前需做妊娠检查以排除妊娠。
育龄女性需在治疗期间和治疗后1个月内确保有效避孕。
男性患者需在治疗期间和治疗后6个月内确保有效避孕。
妊娠
动物研究显示本品具有胚胎和胎儿毒性,尚无本品对孕妇影响的临床研究。
不建议在妊娠期间使用本品。
哺乳
尚不了解本品及其代谢物是否可经人乳分泌。由于对胎儿的潜在风险,应建议母亲在接受本品治疗过程中避免母乳喂养。
【儿童用药】
尚无本品用于18岁以下儿童或青少年患者的临床资料,故本品用于18岁以下儿童和青少年的安全性和有效性尚不明确。
【老年用药】
【药物相互作用】
临床前体外研究显示,赛沃替尼可通过多种代谢酶代谢,主要包括CYP1A2、CYP3A4和CYP3A5等。临床药代动力学研究中,与伊曲康哇200 mg每日一次(一种强效CYP3A4抑制剂)合并用药不会对本品的暴露量产生临床显著性影响(血浆药物浓度-时间曲线下面积(AUC)和峰浓度(Cmax)增加均小于15%),因此,CYP3A4抑制剂不太可能对本品的暴露量产生显著影响。在另一项临床药代动力学研究中,合并服用利福平(一种强效CYP3A4诱导剂)600 mg每日一次连续8天,会使本品的AUC和Cmax分别降低61%和55%,建议应避免本品和CYP3A4的强诱导剂(如苯妥英、利福平和卡马西平)同时使用。CYP3A4的中度诱导剂(如波生坦、依法韦仑、依曲韦林和莫达非尼) 对本品暴露量的影响情况未知,但同样可能降低本品的暴露量,因此本品也应谨慎或尽可能避免与CYP3A4中度诱导剂合用。对于贯叶连翘(St. John's Wort)及其提取物应在本品服用前3周禁服。目前尚无临床试验证实CYP1A2强效抑制剂和诱导剂对本品的影响,因此本品首次服用前1周内及后续服用本品期间,患者应避免使用强效CYP1A2抑制剂和诱导剂。赛沃替尼及其代谢产物M2和M3对CYP2C8有中等程度的可逆抑制,对CYP3A4/5 和CYP2C9有弱的可逆抑制。临床药代动力学研究中,本品600 mg单次给药与咪达唑仑(一种敏感的CYP3A4/5底物)合用,咪达唑仑暴露量无明显变化(Cmax降低16%, AUC降低小于5%),因此本品可以与为CYP3A4/5底物的药物合用。同时由于本品对 CYP2C9的影响弱于对CYP3A4/5的影响,因此也不太可能与CYP2C9的底物发生药物相互作用。但尚无法排除对CYP2C8敏感或治疗窗窄的底物的影响,因此应谨慎服用敏感或治疗窗窄的CYP2C8底物的药物,并监测其与赛沃替尼合用可能导致这些药物暴露量的增加所带来的安全性风险。
赛沃替尼和M2对多药及毒素外排转运蛋白(MATE) 1和MATE2K有一定的抑制作用,因此应慎用二甲双胍,并监测由于二甲双胍暴露量增加可能带来的风险。
赛沃替尼对P-糖蛋白(P-gp)有弱的抑制作用,因此应慎用敏感的P-gp底物并监测可能导致的P-gp底物暴露量的增加所带来的安全性风险。
赛沃替尼不太可能与 CYP1A2、CYP2A6、CYP2B6、CYP2C19、CYP2D6、CYP2E1、 II相代谢酶如葡萄糖醛酸转移酶(UGT) 1A1和UGT2B7、乳腺癌耐蛋白(BCRP)、有机阴离子转运多肽(OATP) 1B1和1B3、有机阴离子转运体1和3 (OAT1和OAT3)以及有机阳离子转运体2 (OCT2)等的底物发生药物相互作用。
在临床药代动力学研究中,合并给予法莫替丁(40 mg单次给药)并不会对本品的暴露量产生临床相关性影响(Cmax降低21%, AUC降低小于15%) 。本品可与改变胃内pH 值的药物合并使用,无需任何限制。
在健康受试者中进行的全面QT研究显示,单次口服赛沃替尼600 mg,可能引起相关的QTc延长,且与赛沃替尼浓度存在正相的线性关系。因此应谨慎合用其他可能导致 QTc延长的药物,并密切观察可能的安全性风险。
【药物过量】
目前尚不清楚过量服用本品可能产生的危害。本品用药过量没有特定的解毒药。如疑似药物过量,应立即停用本品,对患者进行密切观察,必要时釆取最佳支持治疗。
【临床试验】
MET外显子14跳变的非小细胞肺癌研究2016-504-00CH1是一项在MET外显子14跳变的局部晚期或转移性非小细胞肺癌患者中进行的多中心、单臂、开放的II期研究,评价赛沃替尼单药治疗的疗效、安全性和耐受性。
入组患者为既往接受含伯化疗失败[疾病进展或毒性不耐受]或经医生评估不适合进行标准治疗、MET外显子14跳变且EGFR、ALK、R0S1敏感基因变异阴性、经组织学诊断的局部晚期或转移性非小细胞肺癌(含肺肉瘤样癌和其他非小细胞肺癌)患者。
共70例MET外显子14跳变的非小细胞肺癌患者接受赛沃替尼600 mg或400 mg (依据基线体重)每日一次口服,直至疾病进展或出现不能耐受的毒性。
肿瘤评估标准采用实体瘤疗效评价标准(RECIST) 1.1版,肿瘤评估的时间点为每6 周进行一次;治疗一年后每12周评估一次。
入组的70例患者全部被纳入全分析集。
入组患者的平均年龄(标准差)为68.9 (7.9)岁,其中大部分为高龄(>=65岁的患者占77.1%, >=75岁的患者占22.9%);男性41例(58.6%) ; 28例(40.0%)例患者为 吸烟者;基线时美国东部肿瘤协作组(ECOG)体力状况评分为0分的患者有12例 (17.1%) , 1分的有57例(81.4%) , 3分的有1例(1.4%);疾病分期为IV期的患者 有65例(92.9%),其余5例(7.1%)均为III期;病理诊断为肺肉瘤样癌的患者有25例 (35.7%),其他非小细胞肺癌的患者为45例(64.3%) ; 42例(60.0%)患者既往接受过针对晚期肿瘤的抗肿瘤系统药物治疗。
研究的主要疗效终点为独立阅片委员会(IRC)评估的客观缓解率,次要疗效终点包括疾病控制率、无进展生存期、缓解持续时间、起效时间、6个月的无进展生存率和总生存期等。有效性的分析结果见表4。
| 全分析集(N=70) | |
| 最佳客观缓解,n(%) | |
| 完全缓解 | 0 |
| 部分缓解 | 30 (42.9) |
| 全分析集(N=70) | |
| 疾病稳定 | 27 (38.6) |
| 非完全缓解/非疾病进展a | 1 (L4) |
| 疾病进展 | 7 (10.0) |
| 不可评估 | 5(7.1) |
| 客观缓解率(95%置信区间)(%)b | 42.9 (31.1,55.3) |
| 疾病控制率(95%置信区间)(%)b | 82.9 (72.0, 90.8) |
| 缓解持续时间 | |
| 出现客观缓解后发生疾病进展或死亡的患者数,n(%)c | 20 (66.7) |
| 中位数(95%置信区间)(月)d | 8.3 (5.3,16.6) |
| 中位起效时间(95%置信区间)(月)d | 1.4 (1.4,1.5) |
| 无进展生存期 | |
| 发生疾病进展或死亡的患者数,n(%) | 45 (64.3) |
| 中位无进展生存期(95%置信区间)(月)d | 6.8 (4.2, 9.6) |
| 6个月的无进展生存率(95%置信区间)(%) e | 52.0 (38.6, 63.8) |
| 12个月的无进展生存率(95%置信区间)(%)e | 31.9 (20.3,44.2) |
| 总生存期 | |
| 死亡患者数,n(%) | 41 (58.6) |
| 中位总生存期(95%置信区间)(月)d | 12.5 (10.5, 23.6) |
| 12个月的总生存率(95%置信区间)(%)e | 51.1 (38.6, 62.3) |
【规格】
【批准文号】
(1) 100mg: 国药准字H20210026(2) 200mg:国药准字H20210027
【药品本位码】
(1) 100mg: 药品本位码 86982407000015(2) 200mg:药品本位码 86982407000022
【上市许可持有人】
上市许可持有人:和记黄埔医药(上海)有限公司【上市许可持有人地址】
上市许可持有人地址:中国(上海)自由贸易试验区蔡伦路720弄4号【生产单位】
生产单位:上海合全医药有限公司【生产地址】
生产地址: 中国(上海)自由贸易试验区意威路31弄4号 [2]